
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2024;4(1):0002 | https://doi.org/10.47248/hpgg2404010002
Original Research Open Access
Evaluation of ancient DNA imputation: a simulation study
Mariana Escobar-Rodríguez
1,2

 ,
Krishna R. Veeramah
3
,
Krishna R. Veeramah
3
Correspondence: Krishna R. Veeramah
Academic Editor(s): Joshua Akey
Received: Sep 19, 2023 | Accepted: Dec 24, 2023 | Published: Jan 5, 2024
This article belongs to the Special Issue Paleogenomics, ancient DNA, and genomic tales of human history
© 2024 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Escobar-Rodríguez M, Veeramah K. Evaluation of ancient DNA imputation: a simulation study. Hum Popul Genet Genom. 2024; 4(1):0002. https://doi.org/10.47248/hpgg2404010002
Ancient genomic data is becoming increasingly available thanks to recent advances in high-throughput sequencing technologies. Yet, post-mortem degradation of endogenous ancient DNA often results in low depth of coverage and subsequently high levels of genotype missingness and uncertainty. Genotype imputation is a potential strategy for increasing the information available in ancient DNA samples and thus improving the power of downstream population genetic analyses. However, the performance of genotype imputation on ancient genomes under different conditions has not yet been fully explored, with all previous work primarily using an empirical approach of downsampling high coverage paleogenomes. While these studies have provided invaluable insights into best practices for imputation, they rely on a fairly limited number of existing high coverage samples with significant temporal and geographical biases. As an alternative, we used a coalescent simulation approach to generate genomes with characteristics of ancient DNA in order to more systematically evaluate the performance of two popular imputation software, BEAGLE and GLIMPSE, under variable divergence times between the target sample and reference haplotypes, as well as different depths of coverage and reference sample size. Our results suggest that for genomes with coverage <=0.1x imputation performance is poor regardless of the strategy employed. Beyond 0.1x coverage imputation is generally improved as the size of the reference panel increases, and imputation accuracy decreases with increasing divergence between target and reference populations. It may thus be preferable to compile a smaller set of less diverged reference samples than a larger more highly diverged dataset. In addition, the imputation accuracy may plateau beyond some level of divergence between the reference and target populations. While accuracy at common variants is similar regardless of divergence time, rarer variants are better imputed on less diverged target samples. Furthermore, both imputation software, but particularly GLIMPSE, overestimate high genotype probability calls, especially at low coverages. Our results provide insight into optimal strategies for ancient genotype imputation under a wide set of scenarios, complementing previous empirical studies based on imputing downsampled high-coverage ancient genomes.
KeywordsPaleogenomics, ancient DNA, genomics, imputation, simulations, population genetics
With the development of high-throughput sequencing technologies, the number of published ancient genomes has increased significantly over the past decade, reaching 10,000 earlier this year [1]. However, post-mortem damage, particularly in unfavorable environmental conditions, usually leads to a major decrease in the quantity and length of endogenous DNA molecules, in addition to often being mixed with contamination from other sources [2–5]. Such conditions typically lead to low library complexity, and thus, low sequence coverage (≥1x). Even with enrichment for specific SNPs [6,7], only a few samples reach a level of coverage to confidently call diploid sites. As a result, population genetic analyses must account for the high levels of genotype uncertainty and missingness exhibited in ancient samples. One potential strategy for increasing the information available in ancient samples is to perform genotype imputation. Imputation has traditionally been used to compensate for the variants that are not directly characterized in genotyping arrays [8]. The idea behind this approach is to use a panel of known reference haplotypes with a dense set of SNPs in order to infer sites in a sample that has been genotyped only at a subset of these SNPs, providing a gain in power for downstream analysis [9]. Imputation has become common practice in the context of medical and population genetics involving modern genomes — particularly in genome-wide association studies (GWAS).
However, the potential for circumventing the low endogenous content in ancient DNA (aDNA) using imputation has not yet been fully explored. Although missing data and low coverage still allow for analyses like Principal Component Analysis (PCA) [10,11] or F-statistics [12] that can utilize “pseudo-haploid” calls, methods that require complete diploid genotypes or haplotypes such as analysis of runs of homozygosity (ROH) [13] and detection of segments that are identical-by-descent (IBD) [14] can only be confidently applied in ancient samples of exceptionally high coverage (recent attention has been paid to developing methods that try to perform similar analyses using lower coverage data, though even some of these rely on imputation in the underlying analytical framework [15,16]). In the case of applying imputation to ancient samples, two main issues arise from low coverage: large numbers of missing sites and substantial under-calling of true heterozygous genotypes. Therefore, it is preferable in such situations to use software such as BEAGLE v4 [17] and GLIMPSE [18] that perform imputation based on a probabilistic representation of the genotypes in the form of genotype likelihoods.
A number of recent high-profile studies have attempted to impute aDNA with coverages as low as 0.1x for downstream analysis that require accurate diploid genomes, such as inference of RoH and IBD and chromosome ancestry painting [19–26]. To measure the confidence and accuracy of imputation on population genetic inferences, researchers have taken high coverage ancient genomes, downsampled them to lower coverage, and compared the imputed genotypes to high quality genotypes in the original samples [19,20,22,23,27]. Depending on the coverage tested, these studies have achieved as high as 99% overall genotype concordance, while lower minor allele frequency variants remain difficult to impute. Other research has explored imputation pipelines for low coverage data with various pre- and post-imputation filters, where genotype likelihoods are updated to genotype probabilities based on a reference panel, and resulting low-confidence genotypes (GP < 0.99) are filtered out prior to imputation [21,28]. While this approach may discard a large number of sites, it results in more high confidence calls and higher genotype (>99%) concordance between high-coverage and downsampled ancient samples. A recent comprehensive study by Sousa da Mota et al. [29] of more than 40 ancient samples as old as 45,000 years from across the world combined downsampling of high coverage genomes down to 0.1x with trio sequencing to further examine biases that might result from imputation using GLIMPSE. Interestingly, they found significantly lower imputation accuracy for ancient African genomes, likely due to underrepresentation in the reference panel used for imputation. In addition, they noted how imputation accuracy of rarer variants is negatively impacted by the age of the sample and increasing divergence between the target sample and the reference panel.
While these previous downsampling-based investigations have proved invaluable for understanding the effects of coverage on imputation on ancient samples, they do not provide a truly unbiased examination of certain relevant variables. The effect and performance of imputation across various demographic scenarios is constrained by the limited availability of high coverage ancient genomes and the ascertainment bias in their geographical distribution and age. In addition, imputation of prehistoric ancient genomes, including those older than 10,000 years is performed by comparison to existing high coverage modern reference panels. However, the effect of the degree of divergence of the ancient sample from the reference panel on imputation accuracy has not yet been robustly quantified. Therefore, in this study we use a coalescent simulation approach to explore how the variation in genome coverage typical of ancient DNA affects imputation performance as a function of divergence from the reference population, with the goal of obtaining insight about convenient strategies to follow when imputing ancient samples, and to determine the appropriateness of imputation as a method for filling-in missing data in ancient samples. By evaluating imputation performance on simulated data rather than downsampled real data as in previous efforts, we can explore a wider range of scenarios in a more systematic manner. This approach could also be applied to populations whose ancient genomes are as yet underrepresented, or to non-human species.
Our general framework is to simulate genomic data using a three-population model:
- Pop1: the reference haplotype population;
- Pop2: the target population that will be imputed (i.e., the “ancient population”) and is diverged d generations from Pop1;
- Pop3: an outgroup population with fixed divergence from Pop1 and Pop2 (Figure 1).

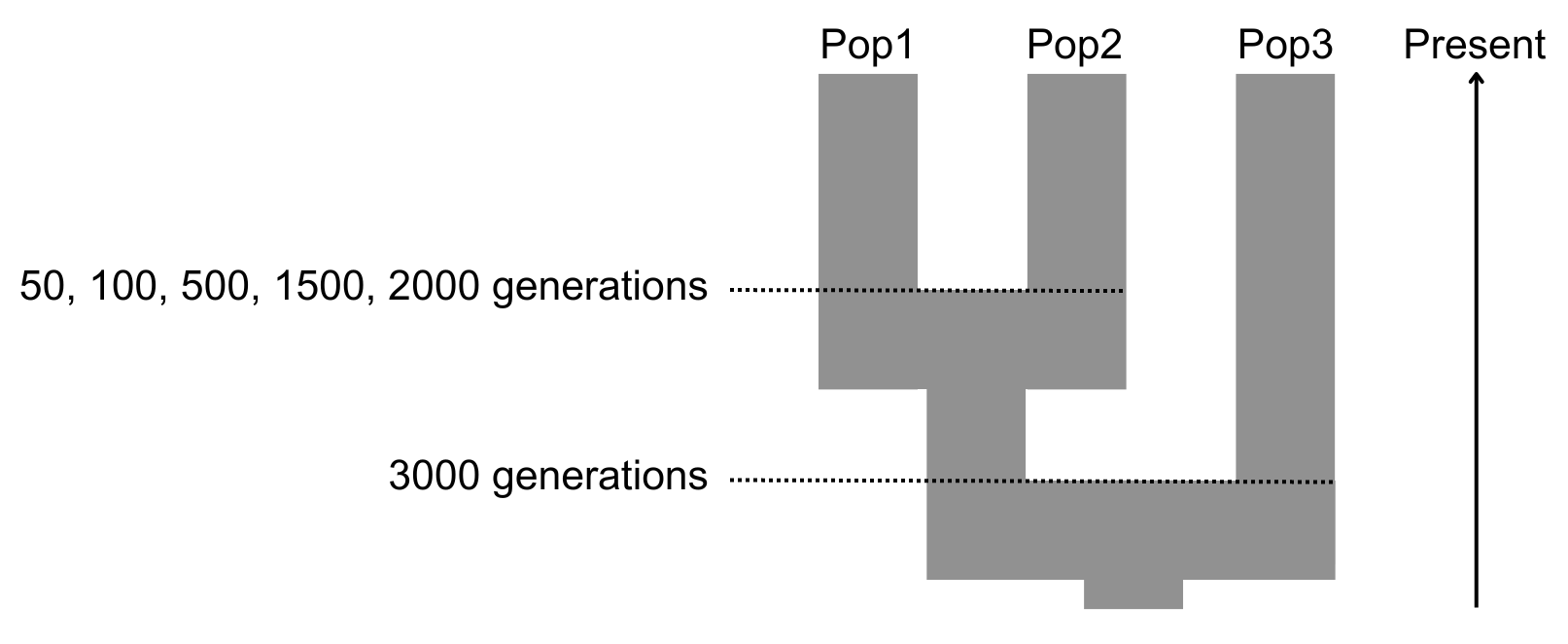
Figure 1 Population demographic model of simulated genomes.
We note that we do not explicitly model an “ancient” Pop2 population that is sampled sometime earlier than Pop1. Instead, we are interested in the effect of the degree of drift between Pop1 and Pop2 on imputation accuracy. Thus, in our model, if present day Pop1 and present day Pop2 diverged 50 generations ago, this would be the equivalent effect of the amount of drift that would occur between present day Pop1 and an ancient Pop2 sampled 50 x 2 = 100 generations ago under a scenario of complete population continuity. This greatly simplifies the parameter space that we are required to explore, as we do not need to consider to what extent ancient Pop2 might have diverged from the lineage from which Pop1 descended before being sampled (of which there are essentially unlimited possibilities). For example, this amount of drift would also be equivalent to both an ancient population sampled 60 generations ago and that diverged from the ancestral population leading to Pop1 80 generations ago, or one sampled 40 generations ago and that diverged 70 years ago.
In order to generate our baseline genomic data (i.e., the truth set), we used fastsimcoal26 [30] to simulate polymorphic haploid genomes roughly equivalent to human chr1 (~220 Mb) using a three-population coalescent model populations under various parameters (Figure 1, Table 1). Effective population size was set to 30,000 for each population. Per site per generation mutation rate was set to 2 x 10–9, and recombination rates were based on the HapMap GRCh37 genetic maps for chr1. Simulated haploid genomes were randomly paired within populations to make diploid individuals. Due to the computational burden when simulating high recombination rates in fastsimcoal26, regions with rates on the order of 1 x 10–7 or above were set to 1 x 10–7. Simulations were performed under five different divergence (d) times for Pop1 and Pop2: d = 50, 100, 500, 1500 and 2000 generations (the last being roughly equivalent to human non-African/African population divergence ~50 thousand years ago assuming 25 years per generation). Pop3 has a fixed divergence of 3,000 generations from the ancestor of Pop1 and Pop2. Our simulations generated ~100,000 biallelic variants above 5% frequency in the reference population, which is within the range expected for current human paleogenomic applications (~400,000 SNPs >5% frequency are found in European populations from the 1KGP based on whole genome shotgun sequencing [31] and 60,000 when filtering down to the most commonly applied 1240K in-solution capture SNP set [6,7]).
Table 1 Simulation parameters.
Furthermore, in order test the impact of a reference panel made up of haplotypes of varying divergence times, we simulated a scenario where the reference panel consisted of N = 1,000 individuals from Pop 1 that diverged, d = 2000 generations from Pop2, while also including five additional individuals that diverged d = 25 generations from Pop2.
In addition, to explore a more complex demographic scenario, we simulated a population expansion in the target population characteristic of the demographic patterns of Mid-Holocene European farmer populations. Using parameters estimated from Veeramah et al. [32] we modeled a population that diverged from the reference population 250 generations ago (~6 Ky) and expanded at a rate of 2% per generation. The target population (Pop2) was sampled 200 generations in the past, resulting in a total of 50 generations of divergence between the target sample and the reference panel (d = 50).
Following simulation by fastsimcoal26, the data from our model were then partitioned and converted into three types of observable datasets: (a) true phased genomes from Pop1 and Pop3 for use as the reference panel for imputation and downstream population genetic analysis; (b) true phased genomes from Pop2; and (c) unphased, low coverage aDNA-like genomes from Pop2. Resulting Arlequin files were converted to VCF files with custom python scripts for all three populations. Low coverage datasets for Pop2 with 0.1x, 0.5x, 1x, and 5x mean coverage were generated from the original true genotypes by first drawing the total number of reads for each site from a random Poisson distribution with γ = 0.1, 0.5, 1, 5, respectively. The number of reads from each allele at true heterozygous sites was then drawn from a binomial distribution with p = 0.5. Finally, to account for sequencing errors, the cycle position of each read was drawn from a random uniform distribution on the interval 1–81 (81 being the total read length and noting that we do not explicitly model variable ancient fragment size) and Phred quality scores were simulated based on an 8th century Viking genome (Figure S1) [22]. We chose not to explicitly introduce aDNA damage, such as deaminations, as the several potential ways to introduce post-mortem DNA damage would have significantly increase the complexity of our data generation, while most common uses cases in paleogenomics largely mitigate such effects through UDG treatment, terminal base clipping, and/or conditioning analyses to transversion sites [2]. SNP calling and genotype likelihood estimation were performed using custom scripts using the expressions described by DePristo et al. [33]. Non-biallelic sites (predominantly triallelic) were removed from any downstream analysis. We note that if a user were to utilize data with aDNA damage, a useful strategy would be to estimate genotype likelihood using methods that incorporate patterns of DNA damage, such as ATLAS [34].
In order to assess how different imputation strategies affect genotype calling accuracy, we examined a number of different of analytical pipelines to impute the ancient-like Pop2. Our strategies differed mainly in the size of reference panels, and choice of imputation software.
Traditionally, most imputation studies for ancient DNA have used large reference panels, such as the 1000 Genomes Project [31]. It has been suggested that rare variants are better imputed by increasing reference size with more diverse populations [35]. By using different reference panel sizes, we aim to shed light on how performance is affected by increasing reference size without necessarily introducing other populations. The reference population sizes used were N = 25, N = 100 and N = 1,000. Additionally, to circumvent the issues that arise with low coverages at fixed genotype calls, we use genotype likelihoods as input to BEAGLE and GLIMPSE, two of the most popular methods for aDNA imputation.
For every tested combination of imputation strategies, divergence times, and coverages, we used genotype likelihoods as input to BEAGLE and GLIMPSE. Phased Pop1 was always used as a reference panel. We imputed the equivalent of chr1 using default imputation window size as defined in BEAGLE v4 [17] and GLIMPSE 1.1.0 [18]. For both software, burn-in iterations were set to 5 (default), and phasing and imputation iterations were set to 10 to increase imputation and phasing accuracy. All other parameters were set to default. Sites with a minor allele frequency (MAF) below 5% in the reference population were removed before imputation, as lower frequency variants are consistently shown to be poorly imputed due to their scarcity in reference panels, which tend to be highly enriched for higher frequency variants. Custom-made genetic maps based on HapMap GRCh37 recombination maps with capping at 1 x 10–7 as described above were used for GLIMPSE imputation, while BEAGLE v4 does not utilize genetic maps. We also briefly compared the results of GLIMPSE 1.1.0 to GLIMPSE v2 [36] with N = 1000, as the latter is optimized for large reference panels with more than 2000 reference haplotypes.
For different combinations of reference and target datasets, we evaluated imputation accuracy by measuring the proportion of matching genotypes between the imputed and the true datasets for Pop2. We also measured accuracy within the context of MAF in the reference population and genotype probabilities (GP). Unless otherwise stated, results are shown for the most recent (d = 50) and the most divergent (d = 2000) reference panels made up of N = 1000 samples to evaluate the accuracy of imputation at our lower and upper bounds. We also examined the potential effect of imputation strategies on population genetic inference using principal component analysis (PCA) via smartpca [10,37], with Pop2 true and imputed genotypes projected onto PC space determined from Pop1 and Pop3 with the parameters lsqproject: YES and no outlier removal (outliermode: 2). Euclidean distance between true and downsampled target individuals was calculated using PC1 and PC2, and scaling the latter by the relative size of the eigenvectors (i.e., percentage of variation explained by each PC). We also explored the accuracy of phasing haplotype in the target Pop2 individuals with the vcftools [38] function diff-switch-error, which assesses the switch error between the true genotypes and those phased by BEAGLE and GLIMPSE.
Figure 2a and Figure S2a summarize the overall proportion of correctly imputed genotypes (our measure of accuracy) in the target population, Pop2, across a range of mean depth of coverages, divergence times (d) from Pop1, and reference population sample sizes (N). In addition to overall concordance, we calculated accuracy separately for true heterozygous sites (Figure 2b, Figure S2b). For samples with a mean depth of coverage of 0.1x overall accuracy for all reference panel sizes and divergence times is within the range of 60%–67%. From 0.5x onwards, the divergence times have a greater effect on accuracy, with more recently diverged target samples being better imputed, as expected due to longer shared IBD stretches between the target and reference samples. This effect is amplified with increase in the reference panel size and coverage, such that the highest accuracies are observed for the smallest divergence (d = 50), largest reference size (N = 1,000) and highest coverage (5x).
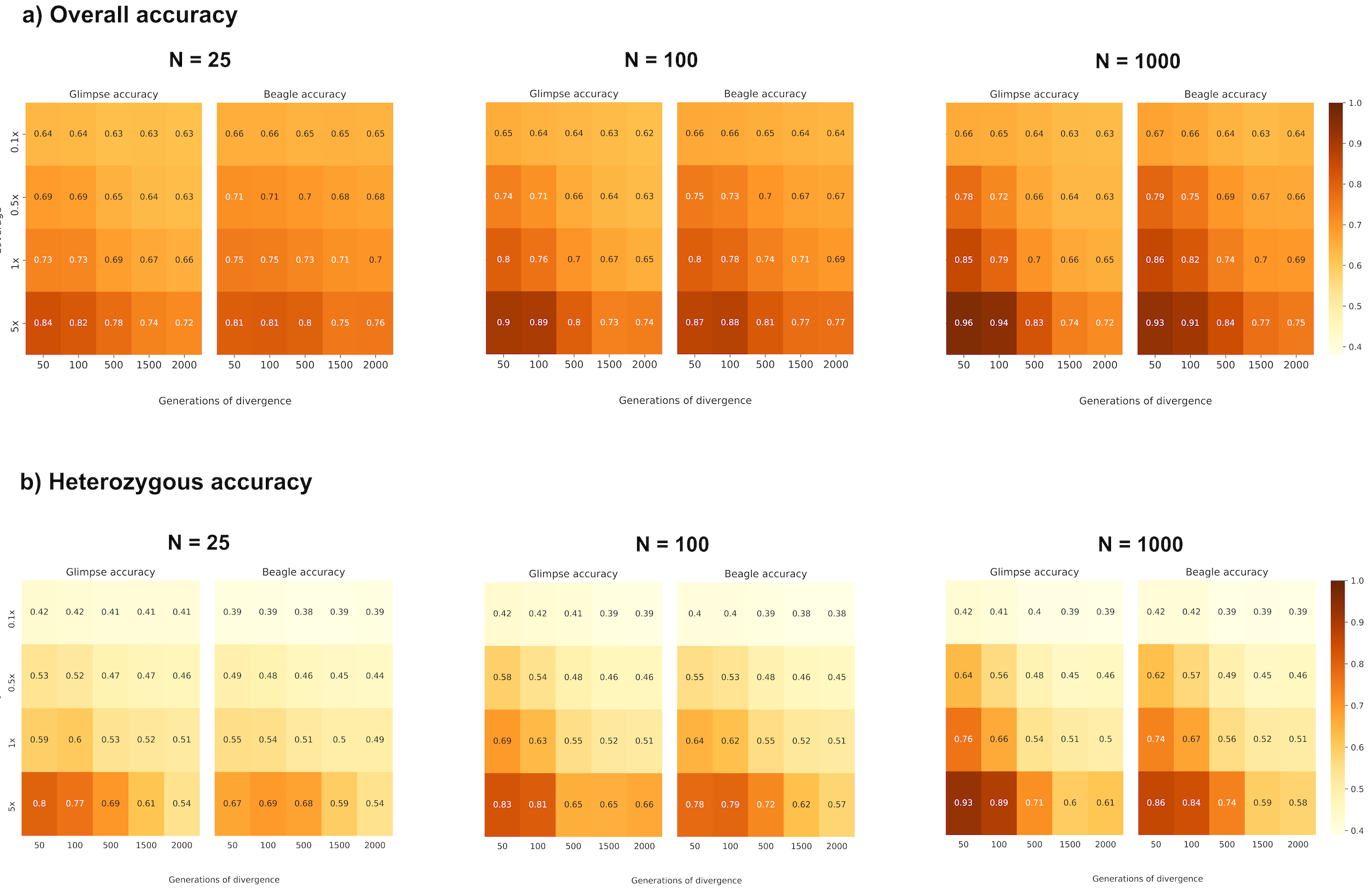
Figure 2 (a) Overall proportion of correctly imputed genotypes; (b) proportion of correctly imputed heterozygous genotypes.
As accuracy at homozygous sites is expected to be higher than at heterozygous sites (especially at lower coverages where often only one sequencing read might be sampled), we also conditioned accuracy at only true heterozygous sites. Heterozygous accuracy is substantially lower than overall accuracy for all combinations of variables, with only the single combination of d = 50, N = 1000 and 5x via GLIMPSE providing somewhat comparable results. At 0.1x, all target sample genotypes have an accuracy of ~40%, regardless of reference population size and divergence time. For 0.5x and above accuracy again predictably increases with coverage and reference panel and decreases with divergence.
Both overall and heterozygous accuracies are similar for d = 1500 compared to d = 2000, perhaps suggesting a saturation point. In addition, when considering all sites, at depths of coverage below 5x, BEAGLE slightly outperforms GLIMPSE, although both software perform similarly. However, at heterozygous sites GLIMPSE is consistently more accurate, especially with increasing coverage and smaller d.
We also note that for all coverages ≥ 0.5x, overall accuracy and accuracy at true heterozygote sizes is consistently higher for small reference sizes and divergence times (N = 25, d = 50) than for a large reference size and divergence time (N = 1,000, d = 2,000). Similarly, while increases in coverage consistently improve accuracy, the more diverged the reference and target, the slower the rate of increase in accuracy that accompanies increases in coverage (across the range of N tested accuracy generally increases by ~15% from 0.5x to 5x for d = 50, but less than 10% for d = 2000). Indeed, greater overall and heterozygous accuracy is consistently achieved when the coverage is 1x and d = 50 compared to when coverage is 5x and d = 1500 or 2000.
Additionally, we compared the performance of GLIMPSE v1 for N = 1,000 to a more recent version of GLIMPSE (v2) optimized for larger reference panels. Both versions of GLIMPSE performed similarly and reached similar levels of accuracy overall and at heterozygous sites (Figure S3).
Finally, we examined a scenario where the primary reference panel (N = 1000) for d = 2,000 was supplemented by five individuals d = 25 generations divergent from the target individual. Despite the addition of these more closely related haplotypes, we observed similar overall and heterozygous imputation accuracy when using a reference N = 1,000 without the individuals of varying divergence (Figure S4).
To examine imputation performance as a function of the allele frequency spectrum, we measured accuracy of imputation genotypes in three bins (0.05-0.1, 0.1-0.3, 0.3-0.5) based on the minor allele frequency (MAF) in the reference population (Figure 3). We report imputation accuracy for the least and most divergent target samples (d = 50 and d = 2000, respectively) to evaluate performance at both upper and lower bounds. We observed that heterozygote imputation accuracy is heavily dependent on MAF, with the best imputed variants being unsurprisingly those in the highest bin (Figure 3b). This effect is particularly pronounced with lower coverage, regardless of imputation method or d. At 0.1x the heterozygote accuracy is ~50% larger for the highest MAF bin (0.3–0.5 ~65%) compared to the least frequent bin (0.05–0.1 < 14%). As coverage increases, rarer variants are generally better imputed. Variants are generally better imputed at d = 50 compared to d = 2000, although the difference in accuracy between both divergence times is again much more evident at rarer variants. While GLIMPSE v1 and GLIMPSE v2 tend to perform similarly overall for N = 1000 for most parameter combinations, when imputing with more divergent reference panels (d = 2000) GLIMPSE v2 did outperform GLIMPSE v1 for the higher frequency variants bin (MAF 10%–50%) at 5x (74% vs. 65%) (Figure S5).
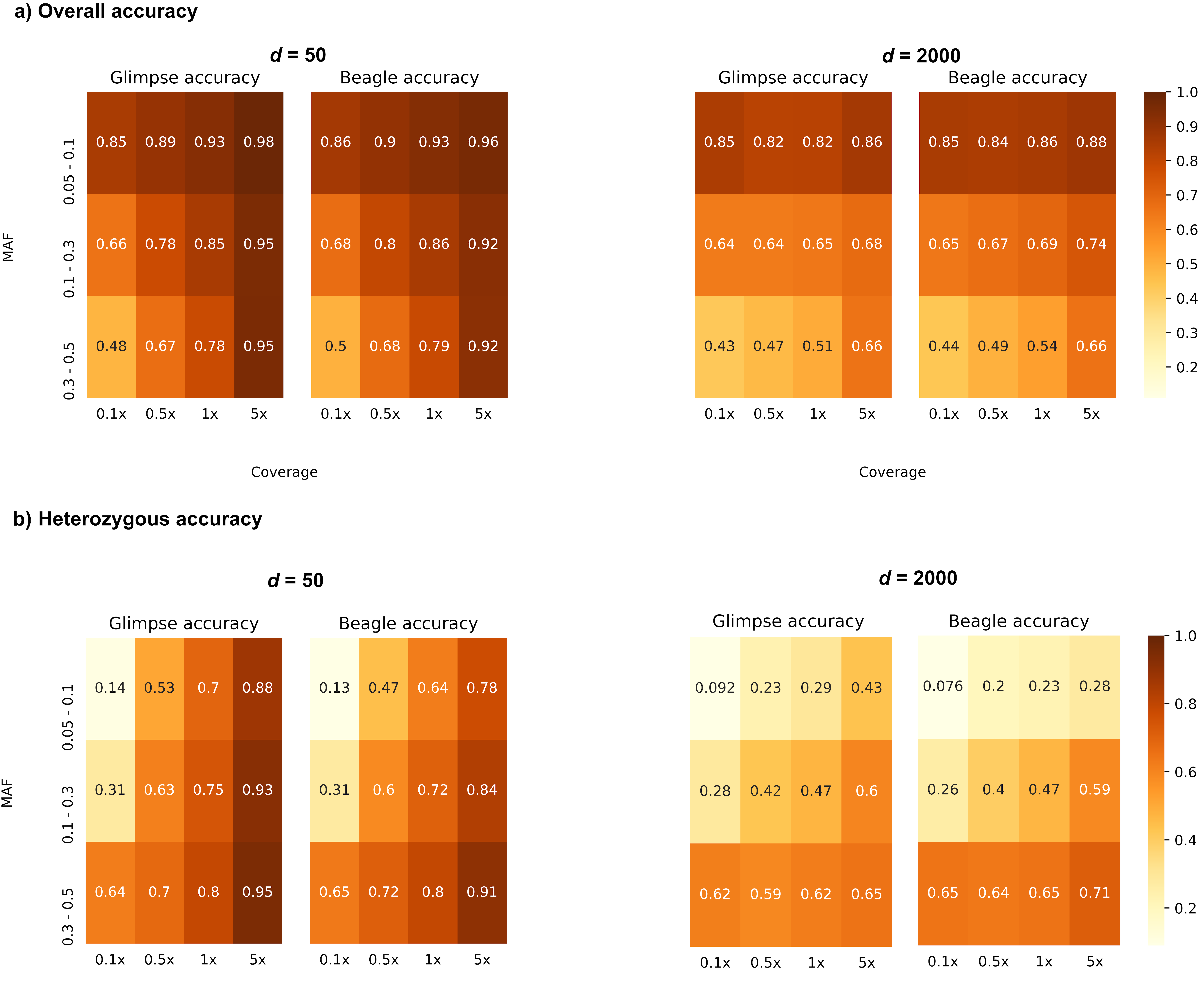
Figure 3 (a) Overall proportion of correctly imputed sites partitioned by MAF in a reference population of N = 1000 and d = 50 generations divergent; (b) proportion of correctly imputed heterozygous sites partitioned by MAF in a reference population of N = 1000 and d = 50 generations divergent.
When evaluating overall accuracy (Figure 3a), we somewhat surprisingly see the opposite pattern: rare variants are imputed correctly more frequently than common variants. A likely explanation for this seemingly counter intuitive observation is that higher MAFs in the reference panel allow for heterozygous sites in the target sample to be better discriminated from homozygous sites. However, when reference MAFs are low, sites could be essentially randomly imputed as homozygous for the major allele and be correct often simply by chance. Such concordance will affect overall imputation accuracy, given that rare variants represent roughly 30% of markers in the reference panel in our simulations (Figure S6).
Once missing sites have been imputed, it is important to decide which sites will remain for downstream analyses. One possibility is to filter sites based on the average probability that a genotype call is correct via estimated genotype probabilities (GP) emitted by the imputation software and restrict analyses to variants with high certainty (e.g., >99%). We examined to what extent these emitted genotype probabilities reflected true error rates (e.g., are sites with a 99% genotype probability correctly imputed 99 times out of a 100?). To avoid the scenario of homozygous variants correctly imputed by chance observed above as a function of MAF, we restrict results to true heterozygous sites. Figure 4 summarizes the proportion of correctly imputed true heterozygous sites relative to the total number of sites imputed as heterozygous within genotype probability bins for d = 50 and d = 2000. While both methods seem to overestimate confidence in genotype calling at all population sizes and coverages, BEAGLE qualitatively tends to outperform GLIMPSE in assigning more representative probabilities for GPs greater than 60%. Although GLIMPSE correctly imputes slightly more heterozygous sites than BEAGLE (Figure 2), the probabilities associated with genotype calls are much less informative. We note that for d = 2000, the genotype probabilities emitted by GLIMPSE are particularly highly discordant (in the direction of being large overestimates) with genotype accuracy, regardless of coverage. While both versions of GLIMPSE were generally similarly discordant for N = 1000, a notable improvement was observed in assigning representative high (>95%) genotype probabilities when using GLIMPSE v2 with d = 2000 compared to GLIMPSE v1 (Figure S7).
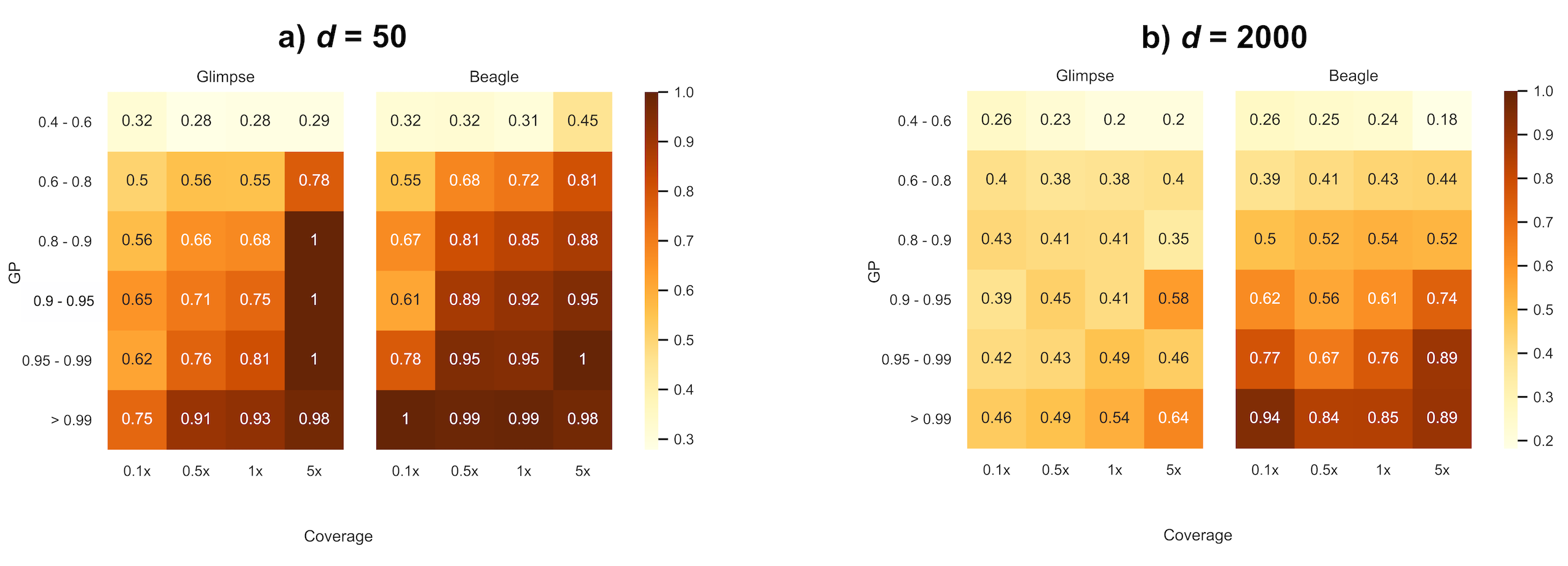
Figure 4 Proportion of correctly imputed heterozygous genotypes partitioned by genotype probability scores (a) d = 50, N = 1000; (b) d = 2000, N = 1000.
Prior to imputation, haplotypes are resolved by assigning non-missing sites to a parental chromosome. As a result, imputation of missing variants will be affected by phasing accuracy. We evaluated phasing accuracy by measuring the proportion of ‘switches’ between the known maternal and paternal haplotypes from the true Pop2 dataset. In Figure 5, we show the proportion of correctly phased sites. Similar to imputation, accuracy is improved when using larger reference panels to phase higher coverage target samples, with a greater accuracy as divergence to the reference panel decreases, and phasing of our most divergent samples does not improve with reference population size, again suggesting a divergence threshold in which haplotype estimation does not improve.
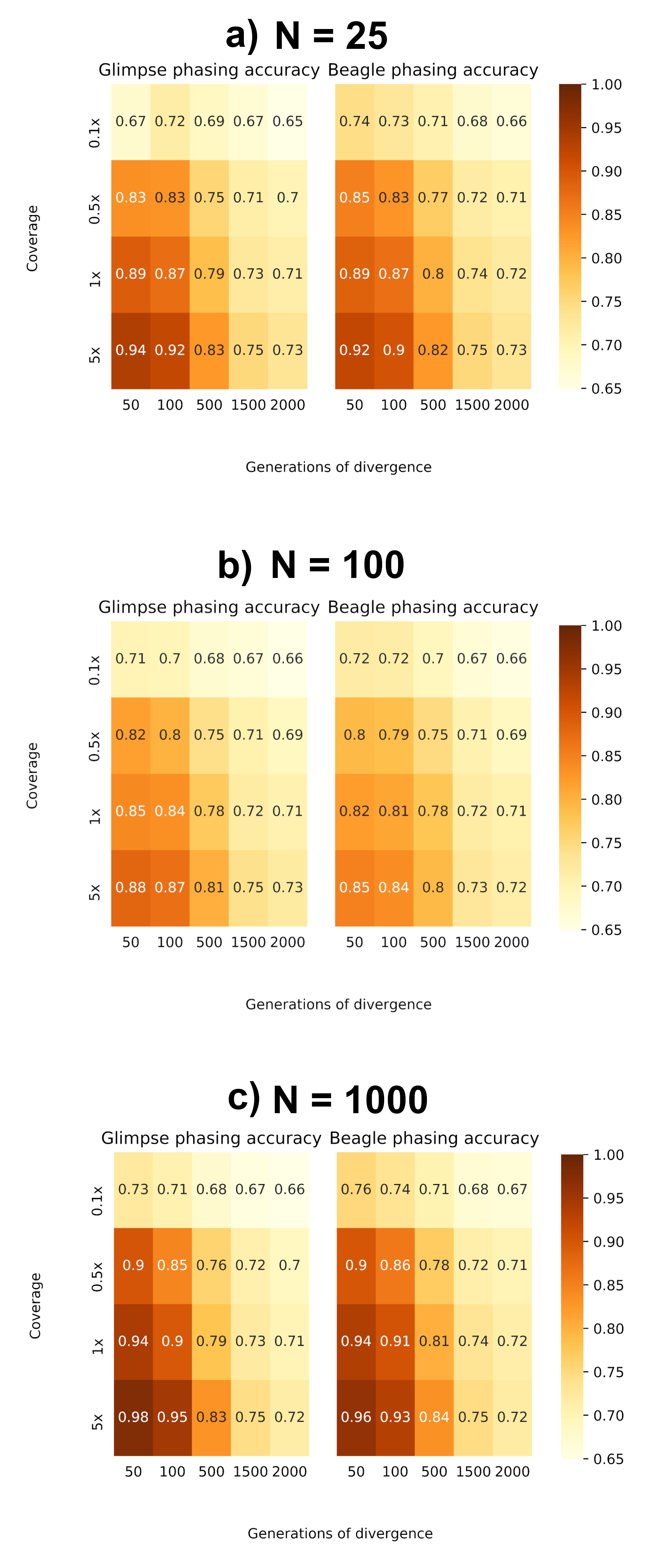
Figure 5 Proportion of correctly phased genotypes based on switch-error. (a) Reference N = 25. (b) Reference N = 100. (c) Reference N = 1000.
In order explore how the kind of discordances between the true genotypes and the imputed genotypes might actually affect downstream analysis, we examined Pop2 imputed samples in a diploid PCA, projecting them on top of Pop1 and Pop3 populations that are 3000 generations diverged (Figure 1) and calculating the Euclidean distance between the true and imputed samples weighted by the relative percentage of variance explained by each principal component (Figure 6). Although we note that PCA can often be performed with pseudo-haploid calls, our analysis focuses on how reliable genotype imputation is for population genomic inference analyses. Figure 6 (outgroup Pop3 not shown for scaling) and Figure S8 show that higher coverage imputed genomes unsurprisingly map more closely to the actual true genotypes and demonstrate smaller variances. Similarly, imputed genomes generally map closer to the reference population as a function of decreasing coverage, with this difference becoming increasingly apparent with increasing d. Noticeably BEAGLE outperforms GLIMPSE for d ≥ 500 in terms of mapping closer to the true population and further from the reference population given the same coverage, but for d ≥ 100 shows the curious pattern of lower coverage samples mapping away from both the reference and true target population and moving towards the outgroup populations, and is thus outperformed by GLIMPSE. Interestingly, imputed samples do not seem to cluster more closely to the true samples when applying post-imputation genotype probabilities filters (GP > 0.99) (Figure S8A) and are systematically at greater Euclidean distances from the true sample than the non-GP filtered samples (Figure S8B). In addition, lower-coverage samples show a more widespread distribution across the PCA when imposing this filter. These observations suggest that genotype probability filtering discards a large number of sites, introducing more noise and without yielding substantial improvements for diploid-based inference.
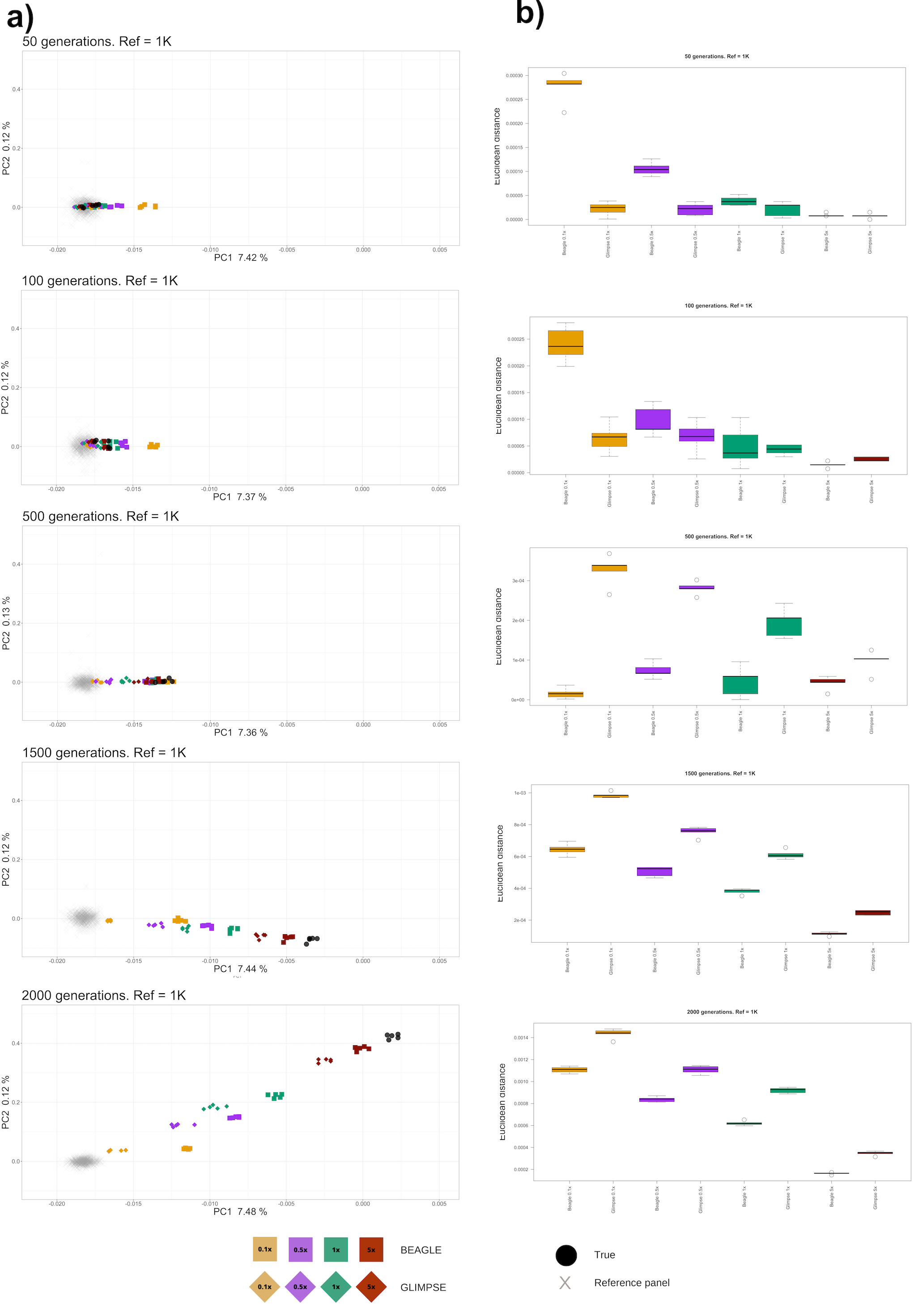
Figure 6 Principal component analysis of imputed and true target samples (outgroup Pop3 not shown). (a) PCA projection; (b) Euclidean distance between true and imputed samples.
While we cannot explore all possible demographic scenarios within the scope of this study, it is generally thought that Eurasian populations grew significantly within the Holocene era. Therefore, to gain some insight into how our results were affected by imposing constant population sizes, we also simulated a scenario in which the target haplotypes originated from a population that underwent population expansion, characteristic of the demographic patterns of Mid-Holocene European farmer populations. We modeled a population that diverged from the reference population 250 generations ago (~6 Ky) and expanded at a rate of 2% per generation [32]. The target population (Pop2) was then sampled 200 generations in the past, resulting in a total of 50 generations of divergence between the target sample and the reference panel (d = 50). Compared to the scenario of d = 50 with no population expansion, we observe a decrease of between 5% to 10% with regard to both overall accuracy of imputation and at true heterozygous sites (Figure S9). When examining this affect across the allele frequency spectrum it is notable that there is a much larger drop off in accuracy for variants in the lower MAF bin under a population expansion (0.05–1, with a difference in error rate of 22% and 24% for GLIMPSE and BEAGLE respectively for 5x) compared to the highest bin (MAF 0.3-0.5, 11% and 2% decrease for GLIMPSE and BEAGLE respectively) (Figure S10). While filtering for SNPs with a MAF > 5% in the reference population might be expected to mitigate most of the potential error introduced by an increase in newer low frequency variants in the expanding target population, there clearly are still residual effects on the haplotype patterns of more established lower frequency variants, likely due to distortion of patterns of linkage disequilibrium.
In this study, we systematically evaluated the performance of imputation of aDNA in different simulated scenarios of population divergence. We measured imputation accuracy in the context of reference MAF, low depths of coverage, reference panel size and imputation software. Unsurprisingly, imputation accuracy is maximized when based on the largest reference panel (N = 1000), the lowest divergence between target and reference population (d = 50) and the highest coverage we attempted (5x). In line with previous downsampling-based studies, accuracy is consistently worse at heterozygous sites and for sites with low minor allele frequencies in the reference populations. However, we note that we never achieve concordance of 99% observed in these same downsampling-based studies (the highest we observe is 96%). This may reflect limitations of our simulated reference panel, which is fairly simplistic compared to the more diverse panels utilized in real-world studies. It is also possible that a larger reference panel such as the Haplotype Reference Consortium (HRC), which combines data from ~32,000 individuals with European ancestry [39] could further improve imputation, especially when combined with new software such as GLIMPSE v2, which is optimized large reference sets such as these. However, it is also possible that the reliance on the assumption in empirical studies that all high-coverage genotype calls are correct may not be completely valid due to the complexity of sequencing errors that occur in next generation sequencing, thus overestimating their accuracy. We would suggest that while our observed accuracy values are not necessarily directly comparable to real-world applications in empirical studies, the general trends in terms of coverage, divergence and software are robust and can be used to guide decision making during imputation.
As would be expected, target samples that were more recently diverged from the reference population were better imputed overall. The greatest impact of increasing divergence on accuracy appears to be at heterozygous sites at low MAF in the reference population. This is in line with the work by Sousa da Mota et al. [29], where imputation of low-frequency variants was negatively impacted by the age and divergence between the African target samples and the reference panel. Interestingly, imputation accuracies for the most diverged target samples (i.e., oldest) we simulated (d = 1500 and d = 2000) were usually within the same range and behaved similarly across every imputation strategy and coverage, suggesting a possible divergence threshold in which imputation performance will not significantly improve with sequencing read depth. We also found that when divergence between the reference haplotypes and target genome was high (d ≥ 1500), this could significantly affect accuracy even when other conditions such as reference population size and coverage were optimum. For example, our results suggest that when assembling reference haplotypes sets, smaller numbers of samples that are more closely related to the target population to be imputed may be a more favorable strategy compared to compiling large numbers of very highly diverged samples. This was also the case even when integrating a few less divergent reference haplotypes into larger and more divergent reference panels. In addition, imputation may perform poorly even in samples with good (in the context of most existing ancient genomes) coverage if the reference sample is distantly diverged from the target (for example imputing an ancient Africa genome at ~5x using non-African reference haplotypes).
Generally, our results suggest that for humans, imputation with modern populations will likely produce usable results for historical-era and young prehistoric populations (for example those that straddle the Neolithic and Paleolithic era), but caution should be applied to the imputation of ancient genomes from the deep prehistoric era (~50,000 years ago), or that are deeply diverged from the reference populations, such as using non-African populations to impute African paleogenomes.
Even the addition of a small number of high coverage ancient genomes to the reference set may significantly improve imputation performance, particularly for inference of heterozygous genotypes with low MAF, as suggested by Ausmees et al. [27].
While accuracy was benefited by increasing the size of reference panels, improvements in accuracy were less pronounced as coverage simultaneously decreased. Most strikingly, imputation performance for the lowest coverage samples did not considerably change with any of the imputation strategies we used. Samples with 0.1x coverage were consistently imputed with ~40% accuracy at heterozygous sites, or ~60% total accuracy, regardless of the imputation strategy or divergence between the reference and target datasets. This can be an important consideration, given that several ancient samples present low coverage (<1x). While our results suggest that imputation of missing data in very low coverage samples may not reach enough accuracy to perform population genetic analyses that require high SNP density, imposing post-imputation GP filtering has been suggested to be a viable option to discard a large number of incorrect calls. While our observations suggest this might be a viable option when imputing with BEAGLE, GLIMPSE filtering may require an alternative approach, since we found it greatly overestimates high GP calls, especially at lower coverages and high divergence. A possible way to circumvent this issue is that proposed by Hui et al. [28], where a pre-imputation GP filter is imposed, at the expense of discarding a large number of SNPs.
Although both software perform similarly, BEAGLE’s overall accuracy is generally slightly higher than GLIMPSE. Accuracy at heterozygous sites may be a more interesting metric to evaluate imputation performance, given that they are less prone to random agreement and will affect overall imputation accuracy. GLIMPSE v1 and GLIMPSE v2 generally outperform BEAGLE at heterozygous sites, especially for more recent samples with higher coverages. When imputing lower frequency variants, GLIMPSE tends to outperform BEAGLE across every simulated coverage. The choice of which method to use may depend on the interest of the user. For example, one might prefer GLIMPSE if the reference and target datasets are not very distantly diverged. If filtering by GPs is of interest, one should consider that GLIMPSE v1 will likely keep a large number of sites with non-representative GPs. Even computational resources may be of consideration, as GLIMPSE constantly outperformed BEAGLE in terms of running time by several orders of magnitude, especially with larger reference panels [18].
When analyzing our imputed diploid genotypes using PCA, higher coverage samples showed closer mapping to true samples, while lower coverage samples clustered with the reference panel. The disparity between true and imputed samples increased with greater divergence times, especially with GLIMPSE imputation at lower coverages. Even at 5x, imputation may not be a particularly suitable method for generating accurate genome-wide diploid genotypes for downstream population genetic analysis if the target and reference population are too far diverged, and may introduce cryptic biases. Interestingly, applying genotype probability filters did not improve clustering to true samples significantly, while the clustering of lower-coverage samples was more noisy. These results suggest that the common practice of genotype probability filtering may not always yield substantial improvements for downstream diploid inference, and instead introduce greater noise.
In addition, PCA presents somewhat of a baseline for population genetic inferential methods, in that it does not rely on signals that correlate amongst sites (i.e., haplotypes). More sophisticated haplotype-based analysis such as IBD inference and chromosome painting will likely result in even larger biases, as our results suggest divergence between the reference and target population has a significant impact on haplotype switch-error, with both increases in coverage and reference size having very little effect once d is ≥ 1500.
By using simulated genomic data, we were able to explore imputation performance for ancient DNA across a wide range of demographic space with respect to divergence time between the target and reference populations with high confidence in the underlying “truth set”, and make observations that might have practical relevance to those designing imputation experiments for their own data. Yet, it should be noted that we only evaluated performance under a very limited number of strategies and simplistic demographic scenarios. Expanding this simulation approach to more complex demographic scenarios that reflect more realistic processes we know are common in human populations such as admixture and isolation-by-distance is likely to be necessary to more fully investigate imputation performance and provide information that can be used to customize imputation pipelines for populations of varying demographic backgrounds. For example, when simulating a significant recent population expansion reflecting the expansion of early European farmers, we found a notable reduction in accuracy at lower frequency variants compared to our more simplistic scenario of constant effective population sizes. While simulation approaches are unlikely to ever capture the real-world sequence features of paleogenomes that downsampling studies of imputation performance can such as DNA damage, they may prove to be a useful complement, particularly for those working on human paleogenomes from underrepresented regions as well as non-human species.
Figures S1 – Figure S10 can be downloaded at: HPGG2404010002SupplementaryMaterials.zip.
Not applicable.
Not applicable.
Krishna R. Veeramah is a member of the Editorial Board of the journal Human Population Genetics and Genomics. The author was not involved in the journal’s review of or decisions related to this manuscript. The author has declared that no other competing interests exist.
All simulation scripts are available at github.com/marianaer/imputation.
MER and KRV conceived the study and wrote the paper, MER performed the analysis.
| 1. | Callaway E. ‘Truly gobsmacked’: Ancient-human genome count surpasses 10,000. Nature. 2023;617:20. [Google Scholar] [CrossRef] |
| 2. | Orlando L, Allaby R, Skoglund P, Der Sarkissian C, Stockhammer PW, Ávila-Arcos MC, et al. Ancient DNA analysis. Nat Rev Methods Primer. 2021;1:14. [Google Scholar] [CrossRef] |
| 3. | Peyrégne S, Prüfer K. Present-Day DNA Contamination in Ancient DNA Datasets. BioEssays. 2020;42:2000081. [Google Scholar] [CrossRef] |
| 4. | Briggs AW, Stenzel U, Johnson PLF, Green RE, Kelso J, Prüfer K, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci. 2007;104:14616-14621. [Google Scholar] [CrossRef] |
| 5. | Stoneking M, Krause J. Learning about human population history from ancient and modern genomes. Nat Rev Genet. 2011;12:603-614. [Google Scholar] [CrossRef] |
| 6. | Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499-503. [Google Scholar] [CrossRef] |
| 7. | Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Pääbo S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci. 2013;110:2223-2227. [Google Scholar] [CrossRef] |
| 8. | Naj AC. Genotype Imputation in Genome-Wide Association Studies. Curr Protoc Hum Genet. 2019;102:e84. [Google Scholar] [CrossRef] |
| 9. | Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499-511. [Google Scholar] [CrossRef] |
| 10. | Patterson N, Price AL, Reich D. Population Structure and Eigenanalysis. PLOS Genet. 2006;2:e190. [Google Scholar] [CrossRef] |
| 11. | François O, Jay F. Factor analysis of ancient population genomic samples. Nat Commun. 2020;11:4661. [Google Scholar] [CrossRef] |
| 12. | Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient Admixture in Human History. Genetics. 2012;192:1065-1093. [Google Scholar] [CrossRef] |
| 13. | Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43-49. [Google Scholar] [CrossRef] |
| 14. | Ferrando-Bernal M, Morcillo-Suarez C, Toni de-Dios T, Gelabert P, Civit S, Díaz-Carvajal A, et al. Mapping co-ancestry connections between the genome of a Medieval individual and modern Europeans. Sci Rep. 2020;10:6843. [Google Scholar] [CrossRef] |
| 15. | Ringbauer H, Huang Y, Akbari A, Mallick S, Patterson N, Reich D. Accurate detection of identity-by-descent segments in human ancient DNA. Nat Genet. 2023. [Google Scholar] [CrossRef] |
| 16. | Ringbauer H, Novembre J, Steinrücken M. Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nat Commun. 2021;12:5425. [Google Scholar] [CrossRef] |
| 17. | Browning SR, Browning BL. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am J Hum Genet. 2007;81:1084-1097. [Google Scholar] [CrossRef] |
| 18. | Rubinacci S, Ribeiro DM, Hofmeister RJ, Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat Genet. 2021;53:120-126. [Google Scholar] [CrossRef] |
| 19. | Gamba C, Jones ER, Teasdale MD, McLaughlin RL, Gonzalez-Fortes G, Mattiangeli V, et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat Commun. 2014;5:5257. [Google Scholar] [CrossRef] |
| 20. | Jones ER, Gonzalez-Fortes G, Connell S, Siska V, Eriksson A, Martiniano R, et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat Commun. 2015;6:8912. [Google Scholar] [CrossRef] |
| 21. | Martiniano R, Cassidy LM, Ó'Maoldúin R, McLaughlin R, Silva NM, Manco L, et al. The population genomics of archaeological transition in west Iberia: Investigation of ancient substructure using imputation and haplotype-based methods. PLOS Genet. 2017;13:e1006852. [Google Scholar] [CrossRef] |
| 22. | Margaryan A, Lawson DJ, Sikora M, Racimo F, Rasmussen S, Moltke I, et al. Population genomics of the Viking world. Nature. 2020;585:390-396. [Google Scholar] [CrossRef] |
| 23. | Antonio ML, Gao Z, Moots HM, Lucci M, Candilio F, Sawyer S, et al. Ancient Rome: A genetic crossroads of Europe and the Mediterranean. Science. 2019;366:708-714. [Google Scholar] [CrossRef] |
| 24. | Clemente F, Unterländer M, Dolgova O, Amorim CEG, Coroado-Santos F, Neuenschwander S, et al. The genomic history of the Aegean palatial civilizations. Cell. 2021;184:2565-2586.e21. [Google Scholar] [CrossRef] |
| 25. | Haber M, Nassar J, Almarri MA, Saupe T, Saag L, Griffith SJ, et al. A Genetic History of the Near East from an aDNA Time Course Sampling Eight Points in the Past 4,000 Years. Am J Hum Genet. 2020;107:149-157. [Google Scholar] [CrossRef] |
| 26. | Saupe T, Montinaro F, Scaggion C, Carrara N, Kivisild T, D'Atanasio E, et al. Ancient genomes reveal structural shifts after the arrival of Steppe-related ancestry in the Italian Peninsula. Curr Biol. 2021;31:2576-2591.e12. [Google Scholar] [CrossRef] |
| 27. | Ausmees K, Sanchez-Quinto F, Jakobsson M, Nettelblad C. An empirical evaluation of genotype imputation of ancient DNA. G3. 2022;12:jkac089. [Google Scholar] [CrossRef] |
| 28. | Hui R, D’Atanasio E, Cassidy LM, Scheib CL, Kivisild T. Evaluating genotype imputation pipeline for ultra-low coverage ancient genomes. Sci Rep. 2020;10:18542. [Google Scholar] [CrossRef] |
| 29. | Sousa da Mota B, Rubinacci S, Cruz Dávalos DI, Amorim CEG, Sikora M, Johannsen NN, et al. Imputation of ancient human genomes. Nat Commun. 2023;14:3660. [Google Scholar] [CrossRef] |
| 30. | Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust Demographic Inference from Genomic and SNP Data. PLOS Genet. 2013;9:e1003905. [Google Scholar] [CrossRef] |
| 31. | 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68-74. [Google Scholar] [CrossRef] |
| 32. | Veeramah KR, Rott A, Groß M, van Dorp L, López S, Kirsanow K, et al. Population genomic analysis of elongated skulls reveals extensive female-biased immigration in Early Medieval Bavaria. Proc Natl Acad Sci. 2018;115:3494-3499. [Google Scholar] [CrossRef] |
| 33. | DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491-498. [Google Scholar] [CrossRef] |
| 34. | Link V, Kousathanas A, Veeramah K, Sell C, Scheu A, Wegmann D. ATLAS: Analysis Tools for Low-depth and Ancient Samples. bioRxiv. 2017. [Google Scholar] [CrossRef] |
| 35. | Bai WY, Zhu XW, Cong PK, Zhang XJ, Richards JB, Zheng HF. Genotype imputation and reference panel: a systematic evaluation on haplotype size and diversity. Brief Bioinform. 2020;21:1806-1817. [Google Scholar] [CrossRef] |
| 36. | Rubinacci S, Hofmeister RJ, Sousa da Mota B, Delaneau O. Imputation of low-coverage sequencing data from 150,119 UK Biobank genomes. Nat Genet. 2023;55:1088-1090. [Google Scholar] [CrossRef] |
| 37. | Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904-909. [Google Scholar] [CrossRef] |
| 38. | Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156-2158. [Google Scholar] [CrossRef] |
| 39. | McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279-1283. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




