
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2025;5(4):0008 | https://doi.org/10.47248/hpgg2505040008
Original Research Open Access
The Quantitative Genetics of Human Disease: 3A Interactions—Correlation in State
Kiana Jodeiry
1,2
 ,
Andrew J. Bass
2,3,†
,
Michael P. Epstein
2,3
,
David J. Cutler
2,3
,
Andrew J. Bass
2,3,†
,
Michael P. Epstein
2,3
,
David J. Cutler
2,3

Correspondence: David J. Cutler
Academic Editor(s): Carina Schlebusch, Lounès Chikhi
Received: Oct 30, 2024 | Accepted: Dec 19, 2025 | Published: Dec 26, 2025
© 2025 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Jodeiry K, Bass AJ, Epstein MP, Cutler DJ. The Quantitative Genetics of Human Disease: 3A Interactions—Correlation in State. Hum Popul Genet Genom. 2025;5(4):0008. https://doi.org/10.47248/hpgg2505040008
The third section of an anticipated four paper series distinguishes two different forms of genetic interactions. In this, the first paper of our discussion on genetic interactions, we describe interactions arising from correlation between genotypic and/or environmental states. In the second paper, we will describe interactions arising from non-additivity between uncorrelated factors (epistasis). We illustrate the ways in which correlations in allelic and genotypic state dramatically alter our intuitions and our quantitative genetic estimates, demonstrating why they must be explicitly accounted for. Departures from Hardy-Weinberg equilibrium are understood as a form of interaction caused by correlations in allelic state within a locus. We review the effects of ancestry on correlation in state within a locus and correlation in state between loci (linkage disequilibrium), with the latter contributing to test statistic inflation in association studies. Relatedness is understood as correlation in allelic state due to recent ancestry. Here we show that population structure, i.e., ancestry, is most simply understood as causing correlation in state between factors, and we demonstrate methods to estimate quantitative genetics quantities while accounting for the correlation in state of alleles induced by complex ancestry.
Keywordsquantitative genetics, human disease, genetic interactions, population structure, linkage disequilibrium
At its essence, the goal of human genetics is to discover genes that causally influence disease initiation or progression. At the genome-wide scale, case-control association studies approach this goal by identifying statistical relationships (i.e., correlations) between single nucleotide polymorphisms (SNPs) and disease status. The simplest possible analytic strategies for performing association studies often assume the data have the property of being identical and independently distributed (although there are many more recent strategies for considering the joint effects of multiple variants in a region on phenotype [1-4]). The simplest approach assumes each random variable (in this context, genotypes at SNPs) is mutually independent, sharing the same probability distribution with other variables, and correlations in the state of genotypes is usually treated as some sort of confounding to be "removed".
When conducting GWAS and fine-mapping association studies, researchers often seek to reduce the influence of factors frequently viewed as artifacts: departures from Hardy-Weinberg Equilibrium (HWE), linkage disequilibrium, population structure, and relatedness among the individuals under study. These factors are usually viewed as departures from model assumptions that can bias test statistics or parameter estimation. While this view is certainly true and often extremely useful, here we will view all of these elements as related manifestations of the same phenomena, correlation in state between genetic factors. Departures from Hardy-Weinberg equilibrium are understood as a form of interaction caused by correlation in allelic state within a locus. Linkage disequilibrium is understood as correlation in allelic state between different loci. Relatedness is understood as correlation in allelic state due to recent ancestry. Complex ancestry over many generations (population structure) is understood from its effects on correlation in state within and between loci. Population structure refers to the presence of subgroups of individuals in the sample under study, such as subgroups with different ancestral backgrounds, who differ systematically across loci in their allele frequencies. In this paper, we demonstrate how variation in ancestry can create correlation in allelic state between every site in the genome, violating the assumption of independence of genetic factors, and acting as the fundamental cause of GWAS test statistic inflation. From our perspective, we will view the effects of population structure on genetic association studies as a form of interaction created by correlations in genotypic state, later contrasting it with non-additive interactions between uncorrelated markers (epistasis) in the second paper of our discussion of interactions [5].
We begin with a reminder [6] of our Kempthorne [7] inspired definition of "effect". The effect of being in any particular state of some factor, or any collection of states, is defined to be the average phenotype of individuals who are in that state or collection of states. The effect of an
We find it important to distinguish correlation in state from non-additivity of independent factors, because the results of these two forms of interaction can be vastly different. Non-additivity among independent factors does not change individual effect sizes, but always increases the total variance. Correlation in state is far more complex and can cause changes in individual state effects (changes in means) as well as increases or even decreases in the total variance relative to the sum of the individual state variances. We first encountered this when examining linkage disequilibrium (LD)—the correlation in genotypic state between two different SNPs—where we observed LD to change the mean effects of individual alleles and make the total genetic variance less than the sum of the individual locus variances [6].
Departures from Hardy-Weinberg equilibrium
The first paper in this series [6] attempted to derive major quantitative genetics results with as few assumptions as possible, yet throughout the entirety of that presentation, one major assumption remained: Hardy-Weinberg Equilibrium (HWE). We chose to embrace HWE because it greatly simplified an already complex presentation, and HWE is required to reach many of the "usual" representations of key results e.g., the additive variance due to a locus is
Here we understand departures from HWE as a form of interaction. It is an interaction caused by correlation in allelic state within a genotype. Conversely, independence in the state of the two alleles in a single genotype will be viewed as the definition of HWE. Notice something subtle. We often call ourselves population geneticists. Inherent in the name is the notion of a population, a group of individuals, and population geneticists intuitively and reflexively think of HWE as a property of a group of individuals. While all this is both useful and true, to better understand the effects of population structure, and ancestry more broadly, it will be helpful to think of and define HWE in terms of individuals. If the states of the two alleles in an individual are independent, then that individual is in HWE, otherwise that individual is not. Over any collection of individuals, if the states of the two alleles are uncorrelated in the collection, then that collection of individuals is in HWE, otherwise the collection is not. This collection need not have any "breeding" relationships in the traditional population genetics sense. A locus in a collection of individuals is in HWE when there is no correlation in allelic state within that locus.
Using the notation from the first paper in this series [6], let
Associate with the random process of picking an individual and picking an allele a Bernoulli random variable
because
If our individual comes from a collection of individuals who all have the same chance of having an
Within this population of individuals defined by their shared allele frequency and covariance in allelic state, we can define our usual measures of genotypic and allelic effects at a locus in accordance with the first paper in this series [6]. Here, the genetic effect
To a population geneticist, the term dominance describes the phenotype of the heterozygote. Locus
In our Kempthorne-inspired interpretation [6], effects are defined as the expectation of phenotype given the context in which they occur, in this case a population with a particular covariance in allelic state. In individuals from a different population with a different covariance in allelic state, the effect of an allele will be different. In this interpretation,
Correlation in state changes everything. A locus that would be completely additive in a population in HWE now has both additive and dominance variance. The total genetic variance of the locus is no longer the sum of the additive and dominance variances. In other words, covariance in allelic state induces dominance variance as well as total interaction variance, the latter of which can be negative. In a population with covariance in allelic state, the additive variance, defined as

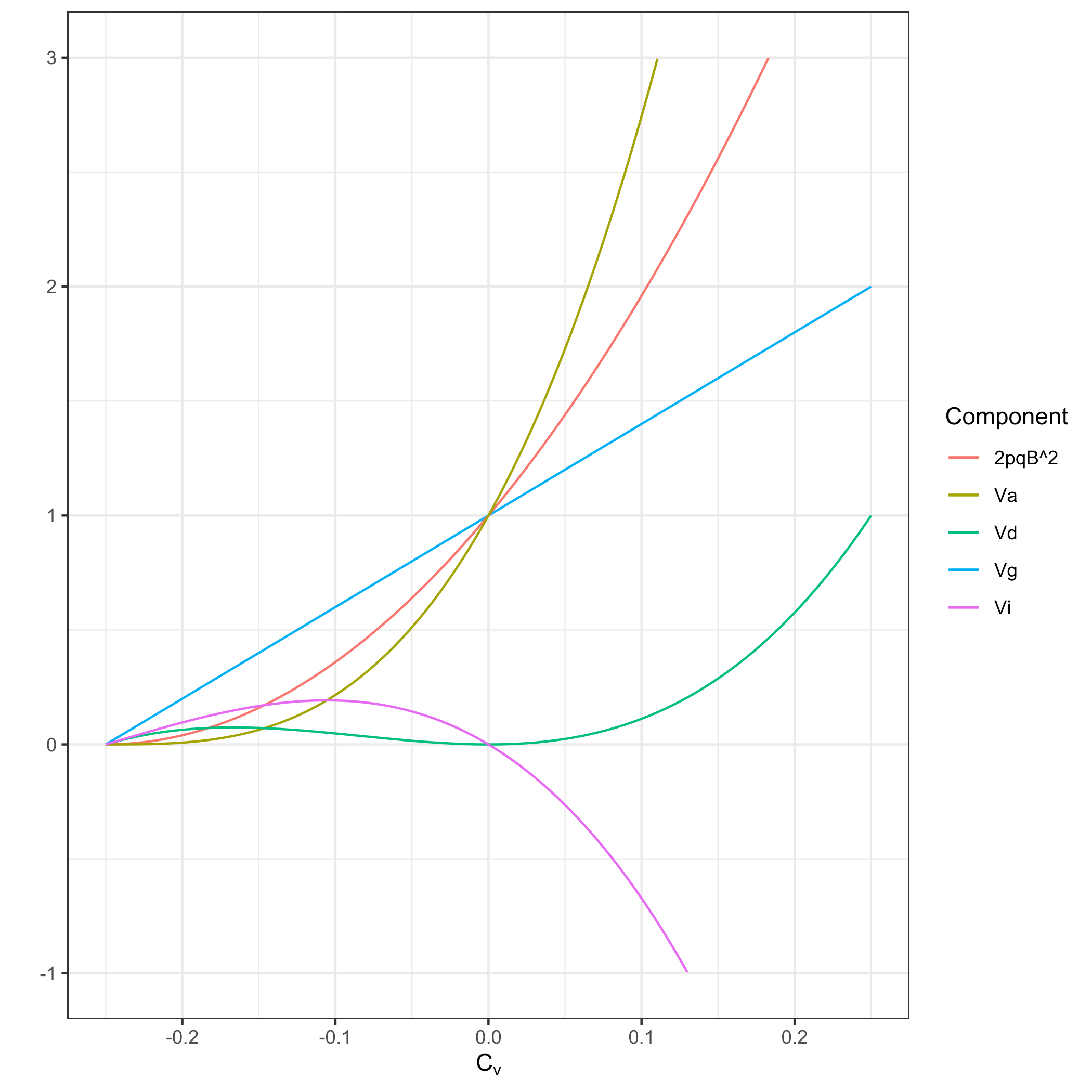
Figure 1 Within locus variance components. Variance components are represented as the ratio of their values in the real population with non-zero covariance in allelic state to their values in an idealized population in HWE, except Vd and VIad which are their values in the real population as they would be 0 in the idealized population.
We will now demonstrate that ancestry can cause correlations in allelic state within a locus that are identified as departures from HWE, as well as correlations in allelic state between loci that are identified as apparent linkage disequilibrium. We additionally show that within-locus correlations in allelic state in a collection of individuals in HWE are a measure of relatedness.
Imagine a collection of individuals who differ in their ancestry. We can think of this collection as a population, but we do not mean to assert anything in particular about breeding within this collection. Pick an individual
Thus, in this collection of individuals, the covariance in allelic state is the average covariance in allelic state within individuals, plus the variance in allele frequency across the ancestries of the individuals in the collection. Notice that we now think of
Since variances are non-negative, we see that absent assortative mating (or some other factor that would create negative covariance in allelic state within individuals), this collection of individuals who vary in their ancestry will have a positive
where
All effect sizes in human genetics should be estimated via a method that accounts for individuals' ancestry - because variation in ancestry creates correlation in state between every site in the genome. This correlation in state between loci caused by variation in ancestry among study participants causes substantial false positive association in genetic association studies if uncorrected. To understand why this is, consider two diploid loci,
Now imagine a collection of individuals whose ancestry differs from one another. Each individual
Note that Equations 38 and 39 follow from the law of total expectation, Equation 44 uses the definition of variance (
We conclude that when a group of individuals varies in ancestry, there will appear to be linkage disequilibrium (covariance in allelic state between loci) at any pair of loci that vary in frequency over ancestry, even if those loci are physically unlinked. From the fact that all correlations are bound between
where
Recall that we assumed
Human population structure can be a major cause of "false positive" association between genetic variation and phenotype whenever there is allele frequency variation in the ancestors of the individuals examined. This has been known for decades, motivating the development of a large range of complementary methods to correct for these effects (e.g., TDT, genomic control, structure, principal component analysis, linear mixed models) [20-24]. To understand more quantitatively exactly what is occurring, imagine attempting to estimate the contribution of locus
where
Now consider the combined effects of all loci
when only a small number of ancestries contribute significantly to variation in allele frequency. This value is necessarily bigger than zero, if there is any variation in
We thus arrive at the full intuition for the need to account for ancestry in human genetic studies. A naive
Patterns common in disease genetics and human demographic history accentuate this inflation. The key aspect to consider is the sign of
In this context, we can best understand how and why various procedures designed to account for this inflation work. Perhaps the most intuitively simple, but complex algorithmically, is the STRUCTURE / STRAT approach developed by Jonathan Pritchard and colleagues [22,26]. The idea is simple, variation in ancestry creates deviations from HWE within loci as well as correlation in allele frequency between loci, therefore, in principle, one can infer ancestral groups from the data [22] and then conditional on the inferred ancestry, perform association studies within ancestry groups in HWE [26]. In the structured association approach developed by Pritchard and colleagues, individuals are assigned to a subpopulation (possibly accounting for admixture with fractional cluster membership) using a model-based clustering program (STRUCTURE) [22] and association statistics are computed stratifying by subpopulation (STRAT) [26]. The approach is natural, but extremely computationally challenging for large sample sizes, and a bit sensitive to a correct a priori determination of the number of "meaningful" ancestral groups contributing to the collection.
At the other end of the computational spectrum is genomic control, pioneered by Devlin and Roeder [20]. Here the approach is incredibly simple, calculate the mean (median)
Since the fundamental problem being solved is caused by covariance in allele frequency and additive variance for the trait, the most natural solution to this problem is to include both in a linear mixed model [24]. Here, the tested model takes the form
where
Linear mixed models can be quite computationally burdensome when compared to a linear model, particularly with thousands of individuals. Note that to perform certain statistical analyses within a linear mixed model framework, such as calculating individual genetic effects, the inverse of the genetic relatedness matrix
Of course, to a geneticist, the most natural way to remove the effects of variation in allele frequency over ancestries is to ensure that the test for association being performed is not affected by correlation in allelic state at unlinked markers. This can this be achieved by focusing only on alleles transmitted from parents known to be heterozygotes [23]. The transmission-disequilibrium test (TDT) counts the number of transmissions from parents known to be heterozygotes to children with some given phenotype, generally a disease state. Because Mendelian segregation is strongly regulated during meiosis to have nearly 50-50 transmission rates, regardless of the identity of any particular allele at a given site, the observed transmission rate from heterozygous parents is a function of the penetrance of the allele for the conditioned phenotype, and this transmission rate is fundamentally independent of any correlations in state at other unlinked sites [31,32]. While this approach is virtually guaranteed to solve issues associated with ancestry, it is often far harder to collect sample sets that include both parents and offspring, and undetected genotyping error can have particularly challenging effects. Computation of the TDT statistic on trios in which one parent is missing marker genotype data increases the type-1 error rate of the statistic [33] as does genotyping error, which can cause apparent over-transmission of common alleles [34,35].
The first paper in this series [6] derived the covariance between individuals in a generalized form. Here we focus on a very specialized case where there is potentially correlation in genotypic state, but no other non-additive interactions of any kind - no dominance, additivity between all loci, and no correlation in state of non-genetic factors. Imagine a collection of individuals that might be a single population in HWE or could be collection of individuals with differing and complex ancestry. We are interested in two randomly picked individuals from this collection; call them individuals
Line 77 uses the lack of dominance or any other interactions extensively. Thus, for an additive trait, the covariance between individuals is the sum over all contributing loci of
If individuals
The covariance in genotypic state at a locus between two individuals is the relatedness between the individuals multiplied by
Thus, if we had a random collection of individuals drawn from a single population in HWE and had a collection of SNP genotypes in those individuals, we could estimate the relatedness between each pair of individuals directly from the genotypes. A matrix
In a collection of individuals with complex ancestries, we again think of an individual's ancestry as determining their personal allele frequency,
Returning attention to the second sum in 80, begin by writing
To finish we need to add the insight that when two individuals are picked at random, if they share no ancestry at a given locus, the allele draws are independent and there is no covariance in state between different loci, so that
Putting this all together with the assumption that local ancestry does not differ significantly across the genome, in other words that for all choices of
Recall that
where
Covariance in genotypic state can therefore be described in several ways. It is relatedness in a collection in HWE. It is also the standard measure of linkage disequilibrium, and variation in ancestry induces LD between sites. Finally it can be the primary "cause" of inflation of test statistics in genetic association studies. Overall, the resemblance between individuals with complex ancestry is a function of
In the two papers that comprise the third part of this series, we distinguish interactions arising from correlations in state between factors from those arising from non-additivity of independent factors.
Correlation in state of factors occurs commonly. Here we understand departures from HWE as a form of interaction caused by correlation in allelic state within a genotype, which can result from variation in allele frequency across ancestry. Correlation in state within loci has very complex impacts, and can cause changes in effects (changes in means) as well as increases or decreases in the total variance relative to the sum of the individual factor variances. In the presence of correlation in state, concepts such as dominance variance or interaction variance are often not well defined, and may appear to be negative, because non-independence of the state of factors can create negative covariance. In the presence of correlations in allelic state, a locus that would have been completely additive had it been in HWE has both additive and dominance variance; its total genetic variance is no longer the sum of the additive and dominance variance; the additive variance is no longer the same as
Variation in ancestry can create correlation in state between every site in the genome. Whenever there is ancestry variation in a collection of examined individuals, any pair of loci that vary in frequency over ancestry will appear to be in linkage disequilibrium, even if those loci are physically unlinked. If there is additive variance for the trait, and if any of the alleles contributing to that variance differ in frequency among the ancestors of the study participants, then there will be test statistic inflation for any naive test correlating allele frequency with phenotype. Correlation in minor allele frequency induced by human demographic history is the primary driver of this inflation in association studies, and we reviewed several approaches to account for this inflation [20,22-24,26].
Correlation is genotypic state between two individuals is also a measure of the relatedness of those individuals. Departures from Hardy-Weinberg Equilibrium (HWE), linkage disequilibrium (LD), population structure, and relatedness are generally viewed by population geneticists as distinct concepts. However, as we have shown, departures from HWE, LD, and relatedness are all measures of the correlation in genotypic state among the individuals in a collection. While it is often useful to treat the effects of each of these independently of one another, all of them can be understood as forms of genetic interaction caused by correlation in state—between alleles within a locus, between pairs of sites across the genome, and between individuals.
Not applicable.
Not applicable.
Not applicable.
This study was supported by NIH Grants RF1 AG071170 and U01 DK134191.
David J. Cutler is a member of the Editorial Board of the journalHuman Population Genetics and Genomics. The author was not involved in the journal’s review of or decisions related to this manuscript. The authors have declared that no other competing interests exist.
All authors participated in the derivation, writing, and editing of this work.
This work has benefited from many helpful suggestions by Greg Gibson, and long conversations with Loic Yengo.
| 1. | de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. [Google Scholar] [CrossRef] |
| 2. | He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9(8):e1003671. [Google Scholar] [CrossRef] |
| 3. | Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82-93. [Google Scholar] [CrossRef] |
| 4. | Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016;32(9):1423-1426. [Google Scholar] [CrossRef] |
| 5. | Jodeiry K, Bass AJ, Epstein MP, Cutler DJ. The Quantitative Genetics of Human Disease: 3b Interactions - Non-Additivity and Missing Heritability. Hum Popul Genet Genom. 2025. [Google Scholar] |
| 6. | Cutler D, Jodeiry K, Bass A, Epstein M. The quantitative genetics of human disease: 1. Foundations. 2023;3(4):0007. [Google Scholar] [CrossRef] |
| 7. | Kempthorne O. The Theoretical Values of Correlations between Relatives in Random Mating Populations. Genetics. 1955;40(2):153-167. [Google Scholar] [CrossRef] |
| 8. | Fisher RA. The Correlation between Relatives on the Supposition of Mendelian Inheritance. Trans R Soc Edinburgh. 1918;52:399. [Google Scholar] |
| 9. | Kempthorne O. The Correlation between Relatives in a Simple Autotetraploid Population. Genetics. 1955;40(2):168-174. [Google Scholar] [CrossRef] |
| 10. | Horner TW, Kempthorne O. The Components of Variance and the Correlations between Relatives in Symmetrical Random Mating Populations. Genetics. 1955;40(3):310-320. [Google Scholar] [CrossRef] |
| 11. | Kempthorne O. The Correlations between Relatives in Inbred Populations. Genetics. 1955;40(5):681-691. [Google Scholar] [CrossRef] |
| 12. | Population Genetics : A Concise Guide. 2nd ed. 2004. [Google Scholar] |
| 13. | Cutler D, Jodeiry K, Bass A, Epstein M. The Quantitative Genetics of Human Disease: 2 Polygenic Risk Scores. Hum Popul Genet Genom 2024;4(3):0008. 2024;4(3):0008. [Google Scholar] [CrossRef] |
| 14. | Wahlund S. ZUSAMMENSETZUNG VON POPULATIONEN UND KORRELATIONSERSCHEINUNGEN VOM STANDPUNKT DER VERERBUNGSLEHRE AUS BETRACHTET. Heredity (Edinb). 1928;11(1):65-106. [Google Scholar] [CrossRef] |
| 15. | WRIGHT S. Genetical structure of populations. Nature. 1950;166(4215):247-249. [Google Scholar] [CrossRef] |
| 16. | Lewontin RC. The Apportionment of Human Diversity. In: Dobzhansky T, Hecht MK, Steere WC, editors. 1972. [Google Scholar] [CrossRef] |
| 17. | Roychoudhury AK, Nei M. Human polymorphic genes: world distribution. New York: Oxford University Press. 1988. [Google Scholar] |
| 18. | Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451(7181):998-1003. [Google Scholar] [CrossRef] |
| 19. | Nei M, Li WH. Linkage disequilibrium in subdivided populations. Genetics. 1973;75(1):213-219. [Google Scholar] [CrossRef] |
| 20. | Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997-1004. [Google Scholar] [CrossRef] |
| 21. | Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904-909. [Google Scholar] [CrossRef] |
| 22. | Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. [Google Scholar] [CrossRef] |
| 23. | Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet. 1993;52(3):506-516. [Google Scholar] |
| 24. | Zhu X, Zhang S, Zhao H, Cooper RS. Association mapping, using a mixture model for complex traits. Genet Epidemiol. 2002;23(2):181-196. [Google Scholar] [CrossRef] |
| 25. | Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J; Schizophrenia Working Group of the Psychiatric Genomics Consortium; et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291-295. [Google Scholar] [CrossRef] |
| 26. | Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;67(1):170-181. [Google Scholar] [CrossRef] |
| 27. | Hao K, Li C, Rosenow C, Wong WH. Detect and adjust for population stratification in population-based association study using genomic control markers: an application of Affymetrix Genechip Human Mapping 10K array. Eur J Hum Genet. 2004;12(12):1001-1006. [Google Scholar] [CrossRef] |
| 28. | Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19(7):807-812. [Google Scholar] [CrossRef] |
| 29. | Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284-290. [Google Scholar] [CrossRef] |
| 30. | Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50(9):1335-1341. [Google Scholar] [CrossRef] |
| 31. | Ewens WJ, Spielman RS. The transmission/disequilibrium test: history, subdivision, and admixture. Am J Hum Genet. 1995;57(2):455-464. [Google Scholar] |
| 32. | Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59(5):983-989. [Google Scholar] |
| 33. | Curtis D, Sham PC. A note on the application of the transmission disequilibrium test when a parent is missing. Am J Hum Genet. 1995;56(3):811-812. [Google Scholar] |
| 34. | Gordon D, Heath SC, Liu X, Ott J. A transmission/disequilibrium test that allows for genotyping errors in the analysis of single-nucleotide polymorphism data. Am J Hum Genet. 2001;69(2):371-880. [Google Scholar] [CrossRef] |
| 35. | Mitchell AA, Cutler DJ, Chakravarti A. Undetected genotyping errors cause apparent overtransmission of common alleles in the transmission/disequilibrium test. Am J Hum Genet. 2003;72(3):598-610. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




