
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2026;6(1):0002 | https://doi.org/10.47248/hpgg2606010002
Original Research Open Access
ArchIE2: A software package for robust inference of introgressed local ancestry
Harold Wang
1

 ,
Sriram Sankararaman
2,3,4
,
Sriram Sankararaman
2,3,4
Correspondence: Harold Wang; Sriram Sankararaman
Academic Editor(s): Joshua Akey, Carina Schlebusch, Torsten Günther
Received: Sep 18, 2025 | Accepted: Dec 13, 2025 | Published: Jan 17, 2026
© 2026 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Wang H, Sankararaman S. ArchIE2: A software package for robust inference of introgressed local ancestry. Hum Popul Genet Genom. 2026;6(1):0002. https://doi.org/10.47248/hpgg2606010002
Introgression is a pervasive feature of human and non-human evolutionary history, and methods that identify introgressed loci have become central to studying its biological impact. We present ArchIE2, an enhanced and more robust version of the reference-free local ancestry framework introduced by ArchIE. ArchIE2 replaces ArchIE’s high-dimensional, sample-size–dependent feature set with a compact collection of summary statistics that generalize across demographic settings. This redesign removes dependency to sample size while preserving predictive accuracy and improving model stability. Across simulations, ArchIE2 matches or exceeds the performance of existing approaches, demonstrating a flexible and scalable framework for detecting introgressed segments, including in scenarios lacking reference genomes from source populations.
Keywordsintrogression, admixture, population genetics, machine learning, human evolution
Introgression—the long-term incorporation of genetic material from one population (the source population) into another (the target population) following admixture—has emerged as an important force in evolution across human [1] and nonhuman species [2,3]. Studies of introgression have been facilitated by the increasing availability of genomic data from extant and ancient populations and the development of analytical methods that attempt to detect various features of introgressed haplotypes. Identifying introgressed DNA segments or loci across individual genomes and studying their properties can provide valuable insights into evolutionary history and biological function [4–6]. A number of methods, such as S* [7], HMM [8], and CRF [9] have been developed to detect introgressed loci across individual genomes (sometimes termed local ancestry inference). Detecting introgressed loci is particularly difficult in ghost introgression, where the source lineage is unsampled. Signals of ancestral structure [10] highlight the importance of approaches that remain effective without explicit source information. Reference-free frameworks address this challenge by enabling inference of introgression from genomic patterns alone.
One approach for inferring ghost introgression uses population genetic statistics that quantify patterns indicative of introgressed tracts. A particularly informative signal is the presence of extended segments of private variants in the admixed population that are absent in a closely related outgroup; this pattern underlies S* [11] and its extensions [7,12,13]. A second class of approaches relies on a generative probabilistic model, such as Hidden Markov Models (HMMs), to model patterns of genetic variation depending on the unobserved state of whether or not a given locus is introgressed [14,15]. A third class of approaches frames the task of detecting introgressed loci as a discriminative machine learning problem where the goal is to directly predict the introgression state based on genomic inputs [16,17].
ArchIE one such discrimative machine learning based method for detecting introgressed segments ghost introgression settings [18]. ArchIE uses a logistic regression model that takes input genomic features to predict the introgression state within a window of an individual’s genome. Although it has shown strong performance in simulations and empirical applications, its feature design poses practical limitations: many of its input features are sample-size dependent, reducing portability, and the high-dimensional, correlated feature space increases the risk of overfitting and complicates model interpretation. To address these issues, we introduce ArchIE2, which preserves the predictive performance of ArchIE while improving its robustness. By condensing the original feature set into carefully selected sample size-independent summary statistics, ArchIE2 enhances the stability of the model while simplifying interpretation.
ArchIE2 is a method to detect introgressed tracts along an individual genome that can be applied in reference-free scenarios. ArchIE2 learns a direct mapping from genomic features to ancestry states, and as such, it requires labeled examples (genotypes of individuals annotated with segments of the genome annotated according to their introgression or ancestry state) for effective training which are generated using population genetic simulators.
The basic training workflow for ArchIE2 is as follows: First, a simulation program generates genotype data under a specified introgression scenario so that the genomes of individuals in the target populations are labeled with their tracts of introgression. Training ArchIE2 consists of two steps. The first step is feature generation, which takes the target and outgroup genotypes (ideally an outgroup that does not share the same introgression event) within each genomic window, combines them with the ground-truth ancestry labels, and produces a feature file for that window. The second step trains the ArchIE2 logistic regression model on this features file. Once trained, the same two-step workflow—feature generation followed by prediction—can be applied to estimate the probability of introgression for each individual genotype within a window. In practice, the demographic history underlying real data may differ from the simulation, but the model is assumed to be robust enough to handle such mismatches and still produce accurate predictions.
The ArchIE2 logistic regression model operates on features extracted from sliding 50kb windows on each haploid chromosome and produces the probability that each genotype within the window is introgressed (Figure 1). The length of 50kb is chosen to be sensitive to archaic introgression events in modern human history (since this is the expected length of introgressed tracts given an admixture time of around 2,000 generations before the present). To generate a SNP-based callset, we run ArchIE2 using a sliding window step size of 10kb, then calculate for each SNP the average probability of archaic ancestry across the five windows that span that SNP.

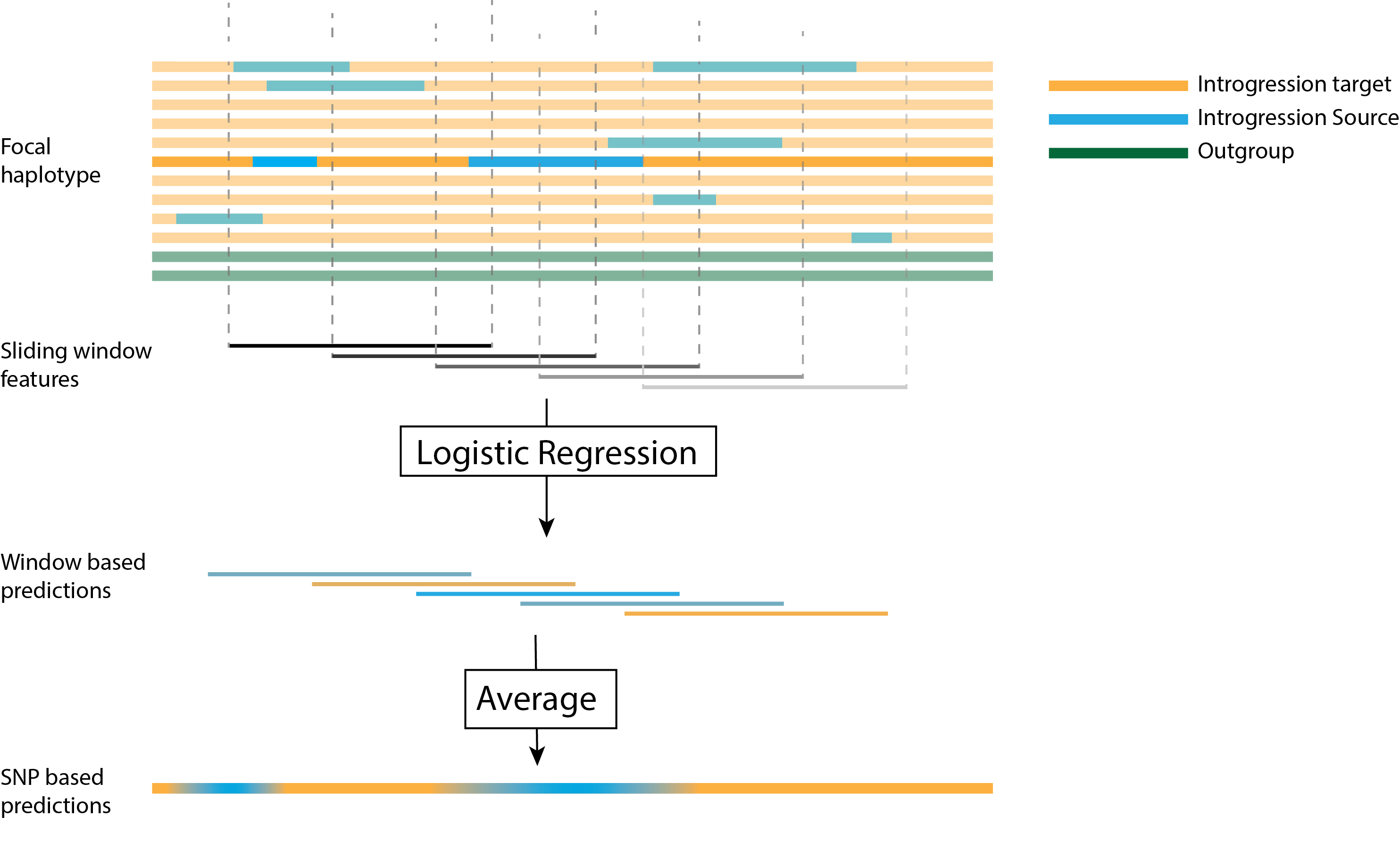
Figure 1. The ArchIE framework. Genetic data are processed into sliding windows, which are then fed into a logistic regression model, the resulting window based predictions are then averaged into SNP based predictions.
Eleven genomic features are used in ArchIE2, falling into two categories: those that require an unadmixed outgroup—a population that does not share the same introgression event as the target population—and those that do not. Among the outgroup-dependent features, ArchIE2 includes the minimum distance between the focal haplotype and the reference population, the number of private SNPs—i.e., SNPs unique to the target population—and the S* statistic. Outgroup-independent features include the first four moments of the distance vector and the first four normalized moments of the individual frequency spectrum (IFS). The first four moments of the distance vector summarizes the length-Ntarget vector representing the Euclidean distance between the focal haplotype and each haplotype in the target population. The normalized first four moments of the IFS summarizes the length-Ntarget vector summarizing the allele frequency spectrum in the focal haplotype (e.g., counts of singletons, doubletons, etc.) (Table 1).
Table 1. Comparison of ArchIE vs ArchIE2 feature set and lengths.
To generate training data, coalescent simulations are performed using msprime [19], which allows the specification of population histories via the Demes format, as well as using a JSON file to specify the sample populations present in the simulation [20]. The script for running msprime simulates introgression from a source population into the target population. It outputs a file detailing for every SNP and sample whether or not it originated in the introgression source population, which we call the SNP based ground truth. For more details, please refer to the github repository (https://github.com/sriramlab/ArchIE2).
We train ArchIE2 on simulations from a specific introgression scenario. In this study, we train the model on the history of Neanderthal introgression [9]. This is a simple scenario that consists of the Neanderthals’ introgression into a European population after the Europeans split off from Africans. For training data, 500 replicates of 10Mb segments are generated, incorporating 50 simulated diploid European samples and 50 African samples. To convert the data for ArchIE2 training, we use sliding windows of length 50kb and step size 50kb, resulting in a cumulative count of 10 million 50kb windows. To obtain ground truth labels, a 50 kb window on a haploid genome is marked archaic if more than 70% of its SNPs are of archaic origin and non-archaic otherwise. The training simulation assumes constant mutation and recombination rates, set by default to 1.25e-8 and 1e-8 per base pair, respectively.
To assess the accuracy of ArchIE2, as well as other methods, we use precision recall curves (PR curves) based on SNP-based callsets. We use PR curves instead of ROC curves due to the imbalanced nature of our dataset, where introgressed sites are rarer than non-introgressed sites. To generate them, we simulate separate testing data in exactly the same manner as in the previous section, with two differences. First, only 100 instead of 500 replicates of 10Mb segments are generated. Second, the sliding window step size is 10kb instead of 50kb for a denser, overlapping coverage. For each scenario, such as different combinations of mutation/recombination rates and different population histories, we generate a separate testing data set. PR analysis was performed based on the SNP callsets generated by the model on the simulated test dataset of interest, compared with the SNP based ground truth.
We developed ArchIE2 as a successor to ArchIE to address two limitations: (i) a dependence on sample-size dependent feature vectors, and (ii) the large number of correlated features, which increased the risk of overfitting and led to unstable coefficient estimates [18]. ArchIE2 addresses these issues by reducing the original high-dimensional set to 11 sample-size independent summary statistics, including the first four moments of the individual frequency spectrum (IFS) and the distance vector, along with features such as minimum distance, the number of private SNPs, and the S* statistic. The mean and variance of the IFS are further normalized to account for sample-size effects (Table 1).
We compared ArchIE2 with ArchIE [18], the S* statistic [7], and the HMMmix [14], using test data simulated under the same demographic history as the training scenario (Figure 2). Condensing the features of ArchIE2 produced virtually no loss in predictive accuracy compared to ArchIE, while yielding a more compact and interpretable model. ArchIE2 also outperformed both S* and the HMMmix. These results indicate that ArchIE2 preserves the predictive strengths of its predecessor while mitigating instability and reducing feature complexity.
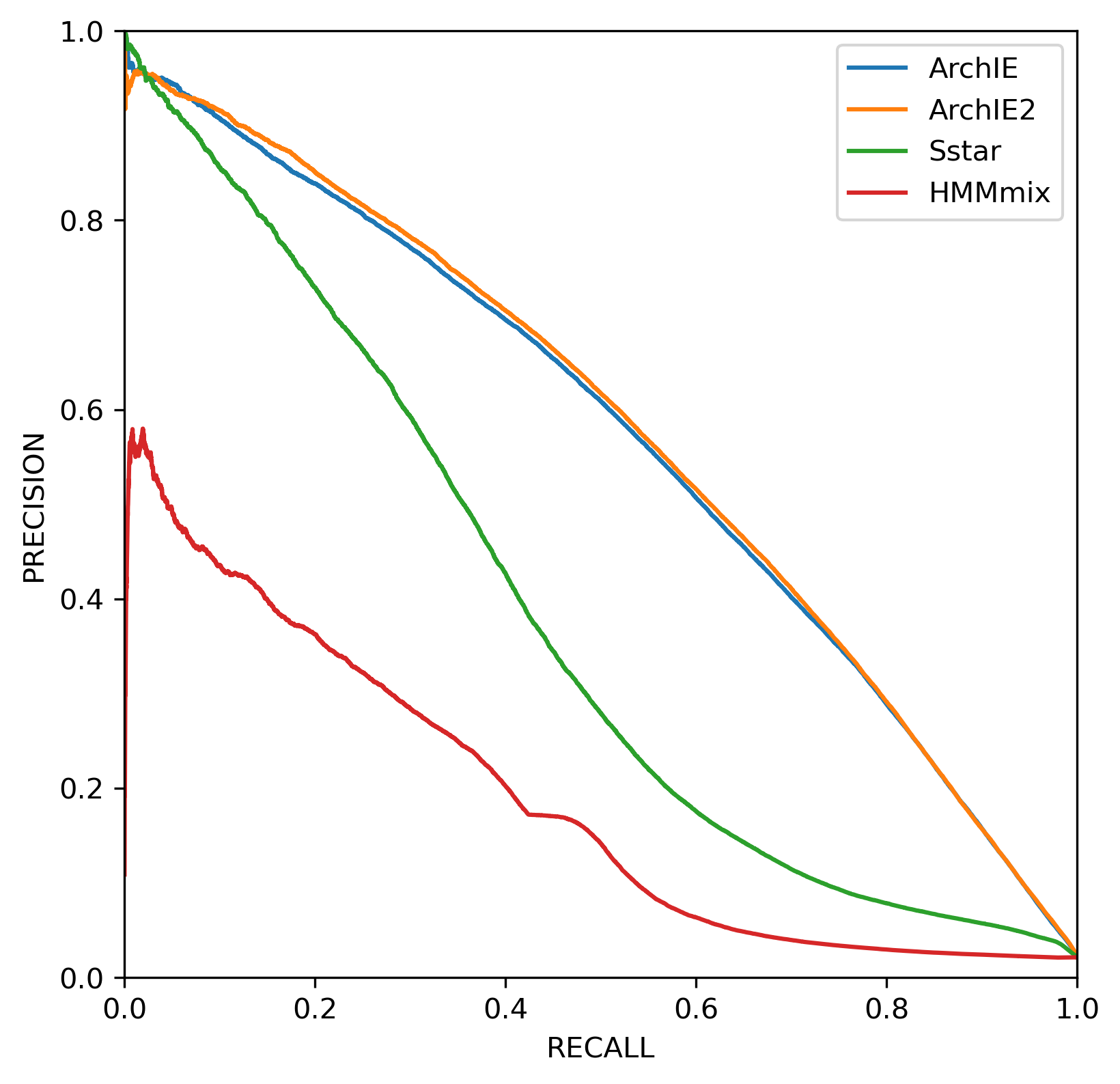
Figure 2. Precision-recall curves for various methods tested on data simulated with μ=1.25e-8.
Here we explore the flexiblity of ArchIE2 over ArchIE with respect to applicability across sample sizes. Here both ArchIE and ArchIE2 models were trained on the base Neanderthal introgression scenario with 100 European haplotypes, but were applied to a test set containing only 30 haplotypes. While ArchIE2 can handle this natively, ArchIE needs to be adapted to this setting. We consider two approaches to adapt ArchIE to this setting: 1) by padding the European genomes in the test set with genomes from a related population, for example Asians, so that the total sample size consists of 100 haplotypes or 2) duplicating genomes to reach the required sample size. ArchIE2 is more accurate than either approach to adapt ArchIE2 (Figure 3) illustrating the value of being able to naturally accommodate varying sample sizes.
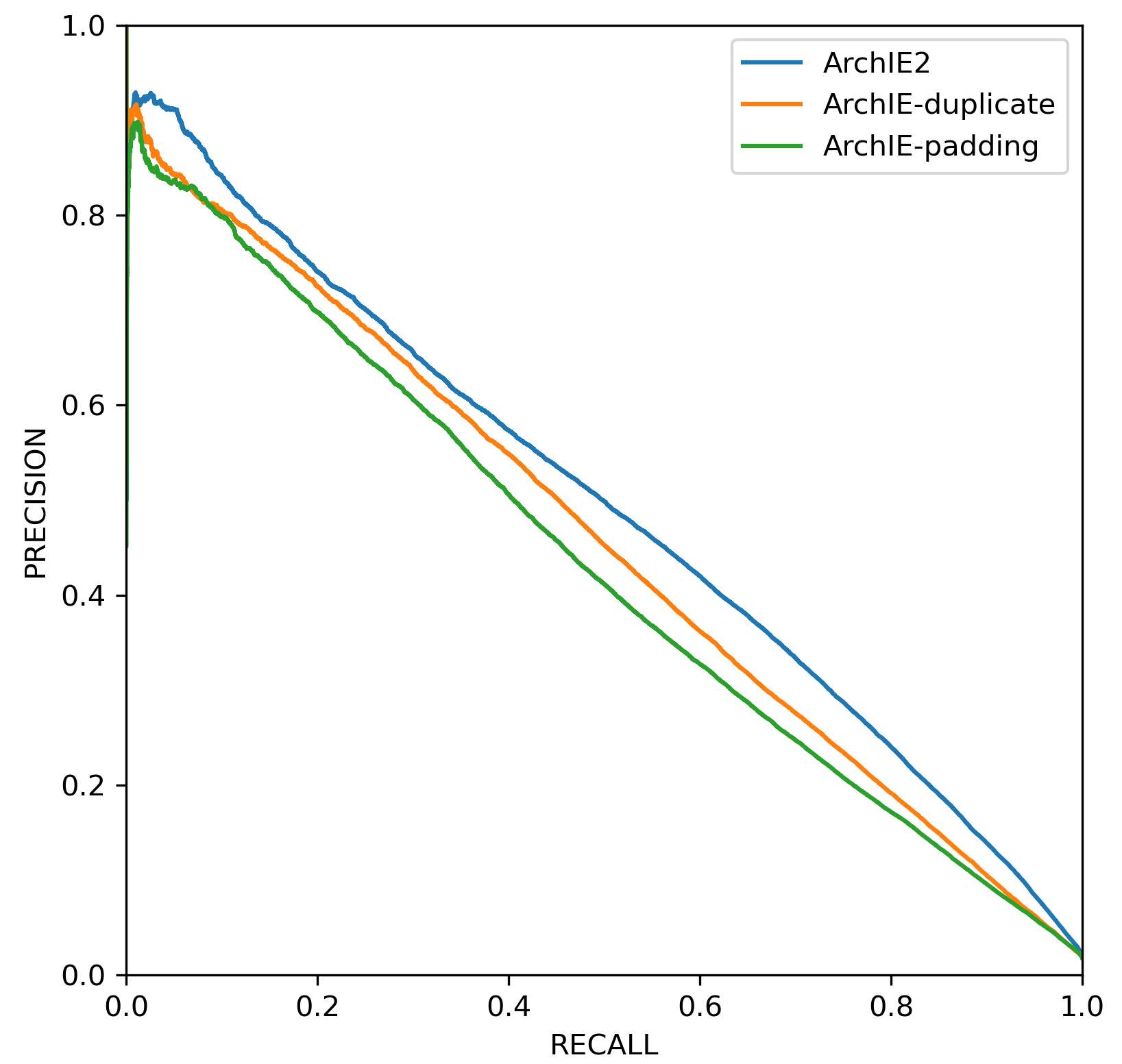
Figure 3. Precision-recall curves for ArchIE and ArchIE2 under mismatch in sample size (100 haplotypes in the training data and 30 haplotypes in the test data).
Given the variation of mutation rates in the human genome, we investigated whether ArchIE2 compares with other methods in response to shifts in mutation and recombination rates. We use the model trained under μ = 1.25e-8, r = 1e-8, and apply them to four scenarios: high vs. low mutation rate and high vs. low recombination rate. In application to real data, we would most probably utilize a single threshold when applying the models to the data. Therefore, we will also report the precision / recall with fixed thresholds for ArchIE and ArchIE2 in Table 2.
Table 2. Precision/recall values across scenarios. One threshold was chosen for each model corresponding to precision=0.8 in the base scenario, and we report its precision/recall in other scenarios while keeping thresholds fixed.
The effects of the change in mutation and recombination rates are consistent between ArchIE and ArchIE2 in Table 2. Low mutation rates and high recombination rates produce precision rates approaching one but very low recall, while high mutation rates and low recombination rates have opposite, albeit less extreme effects. The low recall under conditions of low mutation rates and high recombination rates is likely due to the scarcity of SNPs and the excessively short recombination segments produced.
In this study, we introduced ArchIE2, a scalable and interpretable model for reference-free local archaic ancestry inference. By condensing ArchIE’s high-dimensional, sample-size–dependent feature set into eleven summary statistics, ArchIE2 retains predictive accuracy while improving stability and interpretability. Our benchmarks show that ArchIE2 generally matches or exceeds the performance of existing approaches, including ArchIE, the S* statistic, and HMMmix. In particular, ArchIE2 addresses ArchIE’s practical limitation—most its dependence on fixed sample sizes—by handling varying sample sizes natively, eliminating the need to pad or duplicate samples and the associated loss of accuracy.
Despite these advantages, ArchIE2 has several limitations. Under scenarios with low mutation and recombination rates, the model exhibits miscalibration, likely due to sparse variant density within 50 kb windows; this suggests that window size selection remains an important parameter, and future work could explore adaptive or variable window sizes to improve performance across genomic contexts. ArchIE2 also cannot yet accommodate unphased data, limiting its applicability for certain species or datasets, and extending the framework to handle haploid or unphased genomes would broaden its utility. In addition, evaluating ArchIE2 under ancestral population structure and natural selection remains an important direction for future study. More broadly, integrating additional genomic features and exploring more flexible machine-learning architectures may further enhance the model’s accuracy and adaptability.
Overall, ArchIE2 represents a meaningful step forward in reference-free ancestry inference. Its compact design, robustness under varied conditions, and competitive performance make it a valuable tool for uncovering archaic introgression and advancing our understanding of human evolutionary history.
Not applicable.
Not applicable
No new data were created or analyzed in this study. Data sharing is not applicable to this article
This work was supported by NIH grants GM125055, T32HGT002536, NSF grant CAREER-1943497.
The authors have declared that no competing interests exist.
Conceptualization: H.W. and S.S.; Methodology: H.W. and S.S.; Software: H.W.; Validation: H.W.; Formal Analysis: H.W.; Investigation: H.W.; Resources: H.W.; Data Curation: H.W.; Writing – Original Draft: H.W.; Writing – Review & Editing: H.W. and S.S.; Visualization: H.W. ; Supervision: S.S.; Project Administration: H.W. and S.S.; Funding Acquisition: S.S.
| 1. | Ongaro L, Huerta-Sanchez E. A history of multiple Denisovan introgression events in modern humans. Nat Genet. 2024 Dec;56(12):2612-2622. [Google Scholar] [CrossRef] |
| 2. | Martin Kuhlwilm, Han S, Sousa VC, Excoffier L, Marques-Bonet T. Ancient admixture from an extinct ape lineage into bonobos. Nat Ecol Evol. 2019 Jun;3(6):957-965. [Google Scholar] [CrossRef] |
| 3. | Foote AD, Martin MD, Louis M, Pacheco G, Robertson KM, Sinding MHS, et al. Killer whale genomes reveal a complex history of recurrent admixture and vicariance. Mol Ecol. 2019;28(14):3427-3444. [Google Scholar] [CrossRef] |
| 4. | Huerta-Sánchez E, Jin X, Asan Bianba Z, Peter BM, Vinckenbosch N, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014 Aug;512(7513):194-197. [Google Scholar] [CrossRef] |
| 5. | Taskent RO, Alioglu ND, Fer E, Melike Donertas H, Somel M, Gokcumen O. Variation and Functional Impact of Neanderthal Ancestry in Western Asia. Genome Biol Evol. 2017 Dec;9(12):3516-3524. [Google Scholar] [CrossRef] |
| 6. | Wei X, Robles CR, Pazokitoroudi A, Ganna A, Gusev A, Durvasula A, et al. The lingering effects of Neanderthal introgression on human complex traits. eLife. 2023 Mar;12:e80757. [Google Scholar] [CrossRef] |
| 7. | Vernot B, Tucci S, Kelso J, Schraiber JG, Wolf AB, Gittelman RM, et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science. 2016 Apr;352(6282):235-239. [Google Scholar] [CrossRef] |
| 8. | Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014 Jan;505(7481):43-49. [Google Scholar] [CrossRef] |
| 9. | Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014 Mar;507(7492):354-357. [Google Scholar] [CrossRef] |
| 10. | Cousins T, Scally A, Durbin R. A structured coalescent model reveals deep ancestral structure shared by all modern humans. bioRxiv; 2024. Nat Genet. 2025;57:856-864. [Google Scholar] [CrossRef] |
| 11. | Plagnol V, Wall JD. Possible Ancestral Structure in Human Populations. PLoS Genet. 2006 Jul;2(7):e105. [Google Scholar] [CrossRef] |
| 12. | Vernot B, Akey JM. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014 Feb;343(6174):1017-1021. [Google Scholar] [CrossRef] |
| 13. | Browning SR, Browning BL, Zhou Y, Tucci S, Akey JM. Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell. 2018 Mar;173(1):53-61.e9. [Google Scholar] [CrossRef] |
| 14. | Skov L, Hui R, Shchur V, Hobolth A, Scally A, Schierup M, et al. Detecting archaic introgression using an unadmixed outgroup. PLoS Genet. 2018 Sep 18;14(9):e1007641. [Google Scholar] [CrossRef] |
| 15. | Setter D, Mousset S, Cheng X, Nielsen R, DeGiorgio M, Hermisson J. VolcanoFinder: Genomic scans for adaptive introgression. PLoS Genet. 2020 Jun;16(6):e1008867. [Google Scholar] [CrossRef] |
| 16. | Durvasula A, Sankararaman S. Recovering signals of ghost archaic introgression in African populations. Sci Adv. 2020 Feb;6(7):eaax5097. [Google Scholar] [CrossRef] |
| 17. | Ray DD, Flagel L, Schrider DR. IntroUNET: Identifying introgressed alleles via semantic segmentation. PLoS Genet. 2024 Feb;20(2):e1010657. [Google Scholar] [CrossRef] |
| 18. | Durvasula A, Sankararaman S. A statistical model for reference-free inference of archaic local ancestry. PLoS Genet. 2019 May;15(5):e1008175. [Google Scholar] [CrossRef] |
| 19. | Baumdicker F, Bisschop G, Goldstein D, Gower G, Ragsdale AP, Tsambos G, et al. Efficient ancestry and mutation simulation with msprime 1.0. Genetics. 2022 Mar;220(3):iyab229. [Google Scholar] [CrossRef] |
| 20. | Gower G, Ragsdale AP, Bisschop G, Gutenkunst RN, Hartfield M, Noskova E, et al. Demes: a standard format for demographic models. Genetics. 2022 Nov;222(3):iyac131. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




