1. Introduction
Although the first experimental evidence of cancer stem cells (CSCs) was reported in 1994 by John Dick and colleagues in acute myeloid leukemia [1], it would not be until 13 years later when studies by Li et al. [2] and Hermann et al. [3] published in 2007 would begin to characterize pancreatic CSCs. Over the next decade, hundreds of studies would be published trying to rigidly define and characterize what is a CSC in all solid tumors, including pancreatic cancer. In the early 2000s, seminal studies in the CSC field revealed that, at the onset of tumorigenesis, CSCs formed a distinct population with well-defined traits, such as self-renewal, tumorigenic potential, chemoresistance and metastatic capacity. However, as tumors progressed, “stemness” emerged to be more of a dynamic state or phenotype, rather than a hard-wired entity, which could be acquired in response to environmental pressures during tumor evolution. In line with this more plastic model, in 2017 de Sousa e Melo et al. reported that LGR5-positive CSCs in colorectal cancer were dispensable for tumor progression, as LGR5-negative cells could acquire transient stem-like properties and replenish the LGR5-positive CSC pool if the latter were therapeutically eliminated [4]. Thus, even though the origins of colorectal CSCs may lie within the LGR5-positive intestinal stem cell population [5], other tumor cells can acquire CSC properties over time due to cellular plasticity, temporarily exhibiting stem-like traits, similar to what occurs under homeostatic conditions in the intestinal epithelium [6]. However, in the pancreas, a stem cell with similar characteristics to the intestinal stem cell population has not been described, complicating efforts to define what is a pancreatic CSCs (or the cell of origin). Currently, how we understand stemness, and in particular, pancreatic CSCs, has dramatically changed. The latter has been fueled by the introduction of “omics” technologies and more advanced experimental models, which have provided us with novel insights about the origin and evolution of pancreatic cancer and the roles of different “inner and outer” determinants in these processes, starting with the concept of “stemness” itself. In this review, we provide a comprehensive overview of the intrinsic (genetic and cellular) and extrinsic (microenvironmental and macroenvironmental) factors that govern the emergence, maintenance, and functional diversity of pancreatic CSCs, with a particular focus on the possible origin of these cells, and how these determinants contribute to tumor initiation, progression, and therapeutic resistance.
2. Pancreatic Regeneration and Homeostasis: The Healthy Side
During embryonic development, the pancreas originates from the endoderm. Those cells that start expressing PDX1, or other proposed markers such as NOTCH, are able to differentiate into the exocrine, endocrine and ductal lineages of the pancreas [7, 8]. An extensive review of cell fate during pancreas development and regeneration was published by Stanger and Hebrok [9]. Of those lineages, acinar and ductal cells have been proposed as putative compartments of origin for pancreatic ductal adenocarcinoma (PDAC).
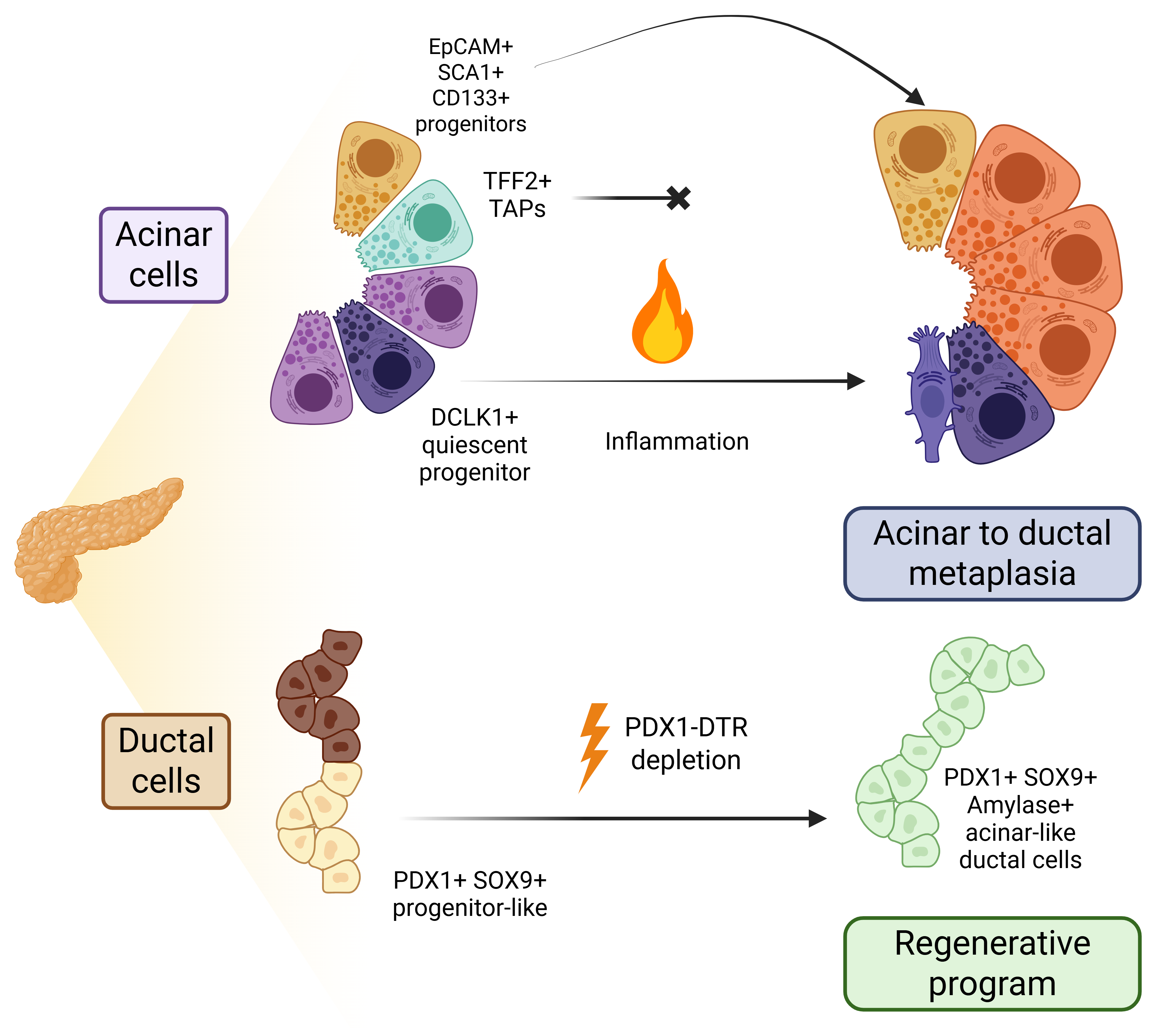
Acinar cells present the highest degree of cellular plasticity in the pancreatic compartment. In response to a damage (e.g., acute inflammation), acinar cells undergo a de-differentiation process called acinar-to-ductal metaplasia (ADM), in which acinar cells lose their polarity and suffer a morphological change towards a phenotype that resembles ductal cells; however, this process does not generate fully differentiated ductal cells [10]. Instead, these cells reside in a hybrid phenotype combining traits of both progenitor cells and some markers attributed to ductal cells (Figure 1). For example, Direnzo et al. indicated that MIST1 is a key factor of mature acinar cell identity, and its loss induces phenotypic changes similar to those observed during ADM [11]. MIST1 defective acinar cells present higher plasticity, form ductal cysts and upregulate markers such as PDX1, HES1, SCA1, FOXA or SOX9 in vitro [12]. Also in vitro, it was proposed that the knockdown of Mist1 could reduce the expression of p21CIP1/WAF1, promoting acinar cell proliferation [13].
Francisco Real and colleagues also presented evidence for the acquisition of a progenitor-like state after culturing acinar cells in non-adherent conditions; however, they did not find an enrichment in ductal markers in vitro. When they applied two different models of pancreatitis in vivo (ductal ligation and cerulein treatment), they observed that the molecular changes were different. While ductal ligation induced the expression of progenitor markers (Sox9, Hes1, Foxa2) as well as Hnf1b and Krt19 (which are associated with ductal cells), cerulein treatment only induced the upregulation of Sox9, and Hnf1b and Krt19 to a lesser extent. Apart from these differences, they also reported a higher induction of senescence (employing β-Gal, p53, p19 and Dec1 as measurements) in the first model. Interestingly, the cerluein model showed an increase in cellular proliferation (12% versus 8% Ki67+ cells), although there was still an increase in p53 and p21, while β-Gal was absent. These results suggest the co-existence of both programmes, proliferation and senescence, depending on the kind of damage (cerulein or ligation-dependent) and suggested a possible heterogeneity in the acinar compartment that regulates different responses to damage [14].
With the development of novel technologies in the last ten years, we now have evidence that the acinar compartment is heterogenous. An interesting study published in 2016 by Wollny et al. employed organoids to study different populations of acinar cells. Among the results they obtained, it is noteworthy to highlight several key observations: a) the authors found that binucleated acinar cells, which exist in homeostatic conditions, do not appear to divide even under inflammatory conditions, unlike mononucleated acinar cells; b) the repopulating capacity upon damage showed by certain acinar cells is transient in time; and c) only a small fraction of acinar cells present regenerative abilities, which they defined as an STMN1-positive population that also expressed the highest levels of SOX9 [15]. Employing in vivo lineage tracing experiments, Westphalen et al. found a rare (0.1–0.5%) population of pancreatic epithelial cells expressing DCLK1, which comprises mostly acinar cells, and that expands in 3D cultures and during tissue damage. These cells present a quiescent phenotype and are present even when there is no tissue damage, suggesting that apart from the increase of progenitor-like cells due to ADM, an adult pancreatic stem cell population could indeed exist [16]. In contrast, a recent article published by Lodestijn et al., using a marker-free lineage tracing model, showed that all acinar cells could contribute to tissue regeneration upon acute injury [17], and there was no clonal expansion when cerulein was administered to mice. More recently, a study led by Jian et al. proposed that DCLK1-positive cells are present in the murine pancreas as a slow-cycling/quiescent population that contributes to long term homeostasis; however, they also identified a TFF2-positive population of transient-amplifying acinar cells, which disappears after injury but can be replenished later [18]. This work contributes to the idea of more than one adult progenitor cell population in the healthy pancreas rather than a possible contribution of all acinar cells during regeneration. In line with this, we have also identified, using the combination of the surface markers EpCAM, SCA1 and CD133, a sub-population of cells in the murine healthy pancreata that express progenitor markers, has multi-lineage potential and presents extended self-renewal capacity. These cells exist as a rare acinar population and, after malignant transformation, contribute to PDAC tumorigenesis [19]. Also, Salas-Escabillas et al. have shown recently that acinar cells can undergo a transdifferentiation process to Tuft-like cells after prolonged damage, such as chronic pancreatitis [20].
Regarding ductal cells (Figure 1), less research regarding their intrinsic plasticity has been published. However, a study published in 2011 claimed that under certain conditions, ductal cells exhibit enough plasticity to regenerate the pancreas. Criscimanna and Speicher et al. employed a diphtheria toxin model under the control of Pdx1-Cre to deplete most pancreatic cells, but they found that most ductal cells remained untouched and that, after some time, there was a regeneration of the pancreas. An extensive analysis of the cellular dynamics of tissue regeneration found that while ductal cells exhibited the highest proliferation rate after diphtheria toxin cell depletion, some acinar Cre-negative cells could have also participated in the regenerative process. Interestingly, the ductal cells involved in the regenerative process presented a more progenitor-like phenotype with increased expression of Pdx1 and Sox9 in addition to some acinar markers such as Amylase, suggesting the existence of a genetic program, similar to ADM, for ductal cells when required [21]. Employing a Sox9:eGFP mouse model, Meritxell Rovira and colleagues studied the progenitor potential of organoids derived from small to big ducts. In this scenario and using single-cell RNA-sequencing, they described different degrees of cellular heterogeneity depending on the size of the ducts used, differences in Sox9 or Spp1 expression, but not in Hfn1b, and validated that some of the populations could give rise to acinar-like cells, similar to what Criscimanna and Speicher et al. observed [22]. It is important to note that both studies relied on studying isolated ductal cells or scenarios involving unlikely conditions, such as the depletion of most pancreatic cells. As a result, the question of how or if ductal cells contribute to regeneration in the context of disease still remains unanswered.
3. Pancreatic Cancer Stem Cells: A History of Multiple Populations
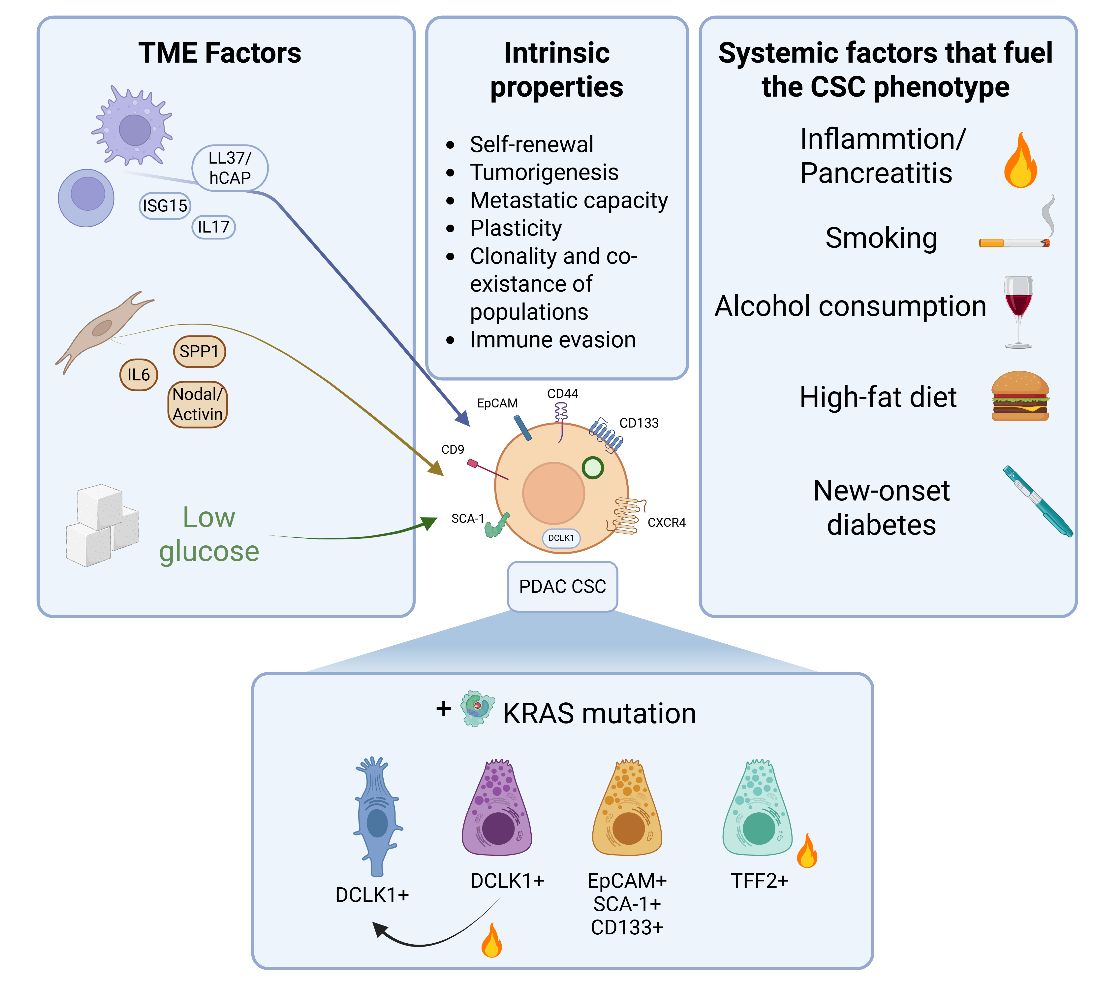
The CSC model has been proposed as an explanation for not only tumor initiation, but also to explain metastases, relapses and therapy resistance. Once believed to be a stochastic model, current evidence defines cancer “stemness” as a state, rather than a hard-wired entity [23]. Li et al. initially described in 2007 that human PDAC cells positive for CD24, CD44 and EpCAM presented 100-fold increase in tumorigenic potential using immunocompromised mouse models and xenografts [2]. Employing the same methodology and additional in vitro and in vivo assays, Hermann et al. showed that CD133-positive PDAC cells presented CSC properties and enhanced chemoresistance to gemcitabine. Also, those cells double positive for CD133 and CXCR4, were identified as highly-metastatic CSCs in vivo [3]. Finally, they also showed that CD133-positive cells were also positive for the markers identified by Li et al. Since then, the number of CSC-associated markers has been expanded with hopes of better identifying this population of cells and linking the CSC compartment to prognosis [24]. Apart from CD24, CD44, CD133 and EpCAM, more surface and intracellular markers in human PDAC cells have been identified over the past 20 years, such as CCR7, cMET, Musashi genes, DCLK1, ALDH1 or CD9 [25-30], along with other functional markers such as riboflavin accumulation in autofluorescent vesicles [31]. For a summary of CSC characteristics and the data presented next, refer to Figure 2. However, the validation of these potential CSC markers has relied on immunocompromised mouse models and patient-derived xenografts, leaving one key question unanswered: "What is the cell of origin of CSCs, and consequently, of the neoplastic cells that give rise to pancreatic cancer?"
With the development of novel genetically-engineered mouse models (GEMMs, summarized in Table 1), the discussion about the cell-of-origin has gained relevance in the PDAC field. The most employed GEMMs to study PDAC were developed to have KRAS activation in embryonic pancreatic cells (Pdx1-Cre; LSL-KrasG12D and Ptf1a-Cre; LSL-KrasG12D) with or without the mutation or deletion of p53 [32, 33]. However, these models induced malignant transformation in a wide variety of cell populations. Since then, multiple efforts have been made to develop PDAC models originating from both ductal and acinar cells. The failure in 2003 to generate tumors in a ductal-based model [34], opened the door to study PDAC tumorigenesis only from an acinar point of view, and thus, pointing to acinar cells as the possible reservoir of cells that upon oncogenic transformation could become CSCs. To restrict tumor initiation to acinar cells in the postnatal stage in a more human-like system, Guerra et al. developed the knocked-in Kras-LSLG12Vgeo allele with double transgenic mice (Elas-tTA; Tet-O-Cre), where the Cre recombinase is under the control of the Elastase promoter and its expression is controlled using an inducible Tet-Off strategy [35]. More recently, other models to study ductal-derived tumorigenesis have been developed and have shown that, although ductal cells are resistant to KRAS-mediated transformation, the addition of other oncogenic events (i.e., p53 loss), generated PDAC [36]. Also, a MSI2-MYC model showed that ductal cells could generate tumors upon MYC activation in a KRAS independent manner [37]. However, and as we have seen before, these two populations present heterogeneity, making the generation of these two kinds of models as imperfect as the prenatal models to identify CSCs. As a possible solution, some groups have employed a different approach, generating models specifically designed to study putative CSC populations. For example, after the identification of DCLK1-positive cells as CSCs by Bailey et al. [30], Westphalen et al. [16] generated a model to trace DCLK1-expressing cells employing the KC mouse model. In this novel model, they validated the quiescent nature of this subset of mainly acinar cells and showed that early pancreatic intraepithelial neoplasias (PanINs) present an enrichment in DCLK1-derived cells, but only in the context of inflammation, and these cells still retained their quiescent state. Interestingly, when they compared a mouse model in which mutated KRAS was expressed in all acinar cells (Mist1-KC) with the DCLK1-KC model, they found that at 2 weeks after inflammation, the Mist1-KC model developed PanINs faster than the Dclk1-KC model, but at 4 weeks, Dclk1-KC mice presented the same number of lesions, providing evidence that DCLK1-positive transformed cells would have between 3 to 9 times more efficiency in forming malignant lesions, and supporting that DCLK1-positive acinar cells could be the population of origin of PDAC CSCs, which still retain the expression of this marker. More recently, Maruno et al. employed a different model to study DCLK1-positive cells by in vivo imaging. The authors found that DCLK1-positive cells also presented high levels of other CSC markers like EpCAM and CD44, and validated that the tumor initiating and metastatic capacities of these cells aligned with a CSC phenotype [38]. Howard Crawdford and colleagues have shown that Tuft-like cells arising from acinar cells, which also express DCLK1, act as CSCs in both neoplasia and carcinoma mouse models. During tumor progression, these Tuft-like cells evolve towards a neural-like progenitor population, that is associated with poor overall survival [20]. These results are in line with those published by Burdziak and Alonso-Curbelo et al. who identified, in a very elegant lineage tracing single-cell RNA-sequencing experiment employing both KC and KPC mouse models (Ptf1a-Cre; LSL-KrasG12D, Trp53R172H), three possible cells of origin/CSCs: Nestin+ progenitors, Tff2+ progenitors and neuroendocrine-like progenitors, which were also originated from Tuft-like cells in these models [39]. However, in this work, the origin of these CSCs remained unclear as all cells were classified as “epithelial cells” without distinction between acinar, ductal or other kinds of populations. In our group, we found that the stem-like population identified by EpCAM, SCA-1 and CD133 (i.e., triple-positive) expanded at early times in the KPC mouse model (Pdx1-Cre model) generating the first pancreatic lesions. After testing this population in vitro, in silico and in vivo, we found that triple-positive cells are bona fide CSCs, which also express high levels of other genes related to the CSC phenotype, such as Dclk1. This triple-positive population was very reduced in ADM compared to early PanINs, indicating that they are even more restricted than the DCLK1 population presented by the other groups. Novel to this triple-positive population was the enrichment in gene signatures associated with immune evasion and metastasis, and the overexpression of the antimicrobial protein Peptidoglycan Recognition Protein 1 (PGLYRP1), which mediated this phenotype [19]. Wang and Ferreira et al. also described another CSC population with different metabolic capacities based on glutamine utilization and the combination of CD44 and CD9, putting into relevance that “fine” identification of CSC subpopulations requires the combination of different markers [28]. Nevertheless, these CSCs also expressed more commonly employed markers such as CD44 and EpCAM. This and other works highlight the co-existence of different CSC clones governing tumor progression and evolution, suggesting that not only DCLK1-positive acinar cells are responsible for generating CSCs and subsequently PDAC. A very recent example validating this concept is the work by Jian et al. where they identify TFF2-positive acinar transient amplifying progenitors and show that the acquisition of a KRAS mutation in these cells makes them resistant to inflammatory clearance observed under homeostatic conditions. Under non-inflammatory conditions, TFF2-positive cells control the expansion of DCLK1-positive CSCs and restrict tumor progression. Indeed, depletion of TFF2-positive cells led to the dramatic expansion of DCLK1-positive clones. Once KRAS is mutated, TFF2-positive acinar cells become longer-lived progenitors instead of transient amplifying cells, and after an inflammatory event, they acquire a CSC-like phenotype characterized by the expression of CD133, CD44, CD24 and CXCR4, which expands in a faster fashion compared to DCLK1-positive CSCs [18]. As KRAS mutation seemed to be highly important to the acquisition of the CSC phenotype, mice were treated with MEK inhibitors to target these populations, and they found a significant reduction in the number of neoplastic lesions. Thus, current evidence strongly suggests that different CSC populations with putative different acinar origins co-exist in pancreatic neoplasias to become the cell of origin for PDAC.
4. Outer Determinants of Pancreatic Cancer Initiation: The Microenvironment and the Host Matters
As previously mentioned, inflammation plays a significant role in the initiation of pancreatic cancer. Guerra et al. described in 2007 that the murine adult pancreas is resistant to KRAS transformation. In an inducible model of KrasG12V, they showed that early postnatal induction of the mutant form of KRAS was capable of generating tumor lesions, although with a higher latency compared to the constitutive model. In adult mice, the induction of KRASG12V was incapable of generating these lesions. Only after an inflammatory insult, such as acute pancreatitis induced by cerulein, did the tumorigenic potential of transformed cells increase, and a wide range of neoplasic lesions could be detected [35]. This study introduced two central concepts that, as seen in the previous section, have been validated years later: first, that not all acinar cells were capable of initiating tumorigenesis, and second, that inflammation is essential to trigger tumor aggressiveness. In fact, this inflammatory process induces pathways like the STAT3-signalling pathway, whose inhibition clearly affects tumor initiation [40]. Thanks to these seminal studies, and the work Jian et al., (mentioned above), we now have a better understanding of how an inflammatory insult could potentially drive the expansion of pancreatic CSCs [18]; however, although chemically induced pancreatitis has been a valuable model for studying its role in tumor initiation, the clinical reality is often more complex, with both macro- and microenvironmental signals contributing to the promotion of cancer stemness (Table 2). In the last years, the tumor microenvironment (TME) has gained relevance as a critical player in tumor initiation and progression, being one of the hallmarks of cancer [41]. In this setting, the works developed by others and us have shed light on how the TME influences pancreatic CSCs. For example, macrophages, the main infiltrated immune cell in PDAC, can induce stemness in several ways. A previous publication from our group identified the antimicrobial peptide human cathelicidin hCAP-18/LL37 as a pro-CSC factor secreted by macrophages. This peptide activates the CSC compartment via formyl peptide receptor 2 (FPR2)- and P2X purinoceptor 7 receptor (P2X7R)-dependent mechanisms, and inhibition of these receptors in in vivo models precluded tumor formation [42]. Also, we found that macrophages secrete interferon-stimulated gene 15 (ISG15) in response to CSC-derived IFNβ secretion, which eventually acts in a paracrine loop promoting the activation of the CSC compartment [43]. In addition, IFNβ could also induce the presence of SCA1-positive CSCs in murine models of pancreatic cancer [44]. An extensive review about how macrophages influence pancreatic CSC biology was published by our group in 2016 [45]. Other immune cells, such as T cells, can also promote CSCs. McCallister and colleagues showed that Th17 cells, usually involved in tissue regeneration, promoted pancreatic CSCs through IL17 secretion [46]. Although other studies have shown how macrophage-derived inflammation promotes tumor initiation and aggressiveness [47, 48], how macrophages contribute to the acquisition of malignant traits in PDAC CSCs is still a field to be fully explored.
Fibroblasts, a key component of the tumor stroma and a major contributor to therapeutic failure at the cellular level, can also enhance the expression of CSC-related markers, such as CD44, ALDH1, and ABCG2, in pancreatic cancer cells. In the study by Nallasamy et al., the authors identified SPP1 as a CSC-related factor, which aligned with CD44 and alpha-SMA expression, with a potential effect on stemness, although the effects of its knockdown were only tested in vitro [49]. These results provide support to the data published by Kesh et al. that described how pancreatic stellate cells, one of the precursors of fibroblasts, promote stemness by upregulating IL6 [50]. The Nodal/Activin pathway has also attracted the attention of researchers and has been found to be activated in CSCs, induced by pancreatic stellate cells [51].
Another important factor influencing the evolution of the TME and the behavior of CSCs is nutrient availability, particularly glucose. In general, limited glucose levels can trigger adaptive metabolic changes in both cancer cells and stromal components, promoting a more aggressive and therapy-resistant phenotype. This metabolic stress can also reinforce stemness traits in CSCs, further contributing to tumor progression and poor clinical outcomes. Pancreatic CSCs present metabolic plasticity, which allows them to survive glucose deprivation by increasing oxidative phosphorylation (OXPHOS) [52]. Interestingly, a study by Nimmakayala et al. proposed that different subsets of CSCs present different metabolic plasticity, and this determines key biological features such as chemoresistance or metastatic potential [53]. We found that the above mentioned ISG15 protein is an important effector in mitochondrial renewal and metabolic plasticity through an ubiquitin-like process known as ISGylation [54]. Loss of ISG15 and ISGylation resulted in the accumulation of defective mitochondria in CSCs, increasing the long-term susceptibility of CSCs to metformin, an inhibitor of complex I of the electron transport chain. This is in contrast to what was observed by Sancho et al, where CSCs use metabolic plasticity to overcome metformin treatment [52]]. CSC metabolic plasticity is a biologically relevant property and one that affords CSCs unique advantages, in addition to treatment resistance. For example, we found that culturing PDX-derived pancreatic cancer cell lines in galactose-containing medium in lieu of glucose-containing medium enriches for a metabolically active CSC population by differentially selecting those cells that can efficiently utilize OXPHOS. This OXPHOS-high CSC population expressed a wide variety of CSC markers like CD133, autofluorescence, CD24 or CXCR4, and interestingly, they were more chemoresistant and immune evasive [55]. Based on the above mentioned and other studies linking CSC, the TME and OXPHOS, we have developed approaches to target signaling pathways promoted by macrophages [42] or, more recently, to target the OXPHOS dependency of CSCs with tailored ruthenium complexes [56].
While there is ongoing debate about how various cell types influence the behavior of cancer cells and CSCs, much less attention has been given to the role of the macroenvironment, or the host, in shaping tumor development. Outer determinants such as risk factors or lifestyle habits are now under study. Smoking, the top risk factor for cancer development, has been studied in the context of pancreatic CSCs. For example, Al-Wadei et al., investigated the impact of nicotine on pancreatic CSCs and the underlying mechanisms involved, finding that chronic exposure to nicotine led to different changes, such as the increased expression of nicotinic acetylcholine receptors (nAChRs), the production of epinephrine and norepinephrine, and the activation of the sonic hedgehog (SHH) pathway. They also found that GABA treatment inhibited the nicotine-induced effects, including the activation of SHH signaling and the enhancement of self-renewal and proliferation [57]. A more comprehensive study was published by Hermann and Sancho et al., in 2014, showing the effects of nicotine in different GEMMs and cell lines. Nicotine induced dedifferentiation of acinar cells by activating the AKT-ERK-MYC signaling pathway. This led to the inhibition of the activity of the Gata6 promoter, resulting in the loss of the GATA6 protein and subsequent acinar differentiation, leading to the promotion of an aggressive phenotype, including induction of epithelial-mesenchymal transition, increased numbers of circulating cancer cells, and their dissemination to the liver. Importantly, nicotine-treated pancreatic cells acquired gene expression patterns and functional characteristics of CSCs. Finally, they found that treatment with metformin attenuated the nicotine-induced effects by upregulating GATA6 and promoting differentiation toward an acinar cell program [58]. Another study examined the effects of cigarette smoke extract, nicotine, and nicotine-derived carcinogens (NNN and NNK) on pancreatic cancer cell lines over 80 days. The study also included treatment of the KC mouse model with cigarette smoke extract for 20 weeks. The authors found that cigarette smoke components activated the cholinergic receptor nicotinic alpha 7 subunit (CHRNA7), leading to the activation of mitogen-activated protein kinase 1 (MAPK1) and FOS-like 1 (FOSL1). FOSL1 subsequently bound to the PAF1 promoter, enhancing its expression and promoting stemness. Additionally, the KC murine model presented an enrichment in PAF1 and an increase in tumor burden [59]. More recently, another group identified that NNK not only has a role in promoting stemness, but also chemoresistance, by inducing autophagy in cell lines [60].
Although ethanol consumption is the second-leading risk factor for developing pancreatic cancer, it has been less studied. A study with only in vitro approaches found that human normal ductal cells exposed to long-term ethanol amounts presented an enrichment in genes and markers related to CSCs [61]. Therefore, while smoking and alcohol consumption are well-established risk factors for pancreatic cancer, their specific roles in disease development and in the activation of the CSC compartment still remain poorly or incompletely understood and require further investigation. Similarly, new-onset type II diabetes, a condition increasingly associated with pancreatic cancer [62], should also be explored in experimental models to clarify its role in tumor development and potential mechanistic link with CSC regulation. In addition to these traditional risk factors, the role of the microbiome in PDAC is gaining attention [63], with emerging evidence suggesting it may influence tumor progression and possibly CSC dynamics. Thus, a comprehensive understanding of how these external factors/outer determinants influence CSC-mediated tumorigenesis is essential for identifying novel preventive and therapeutic strategies to combat PDAC.
5. Inner Determinants of Pancreatic Cancer Initiation: It’s (Almost) all About Genetics
While environmental factors contribute to the onset of PDAC, genetic alterations are central to disease initiation and progression, though not as straightforward as once believed. Gone are the days when the acquisition of mutations, such as KRAS variants, was considered sufficient to induce tumorigenesis. In 2003, Guerra et al. demonstrated that systemic activation of the KrasG12V oncogene fails to induce tumors in most tissues, with the effect being limited to the lungs [64]. Later, the same group showed that an inflammatory insult is necessary in the pancreas to facilitate acinar-to-ductal transformation and tumorigenesis. Indeed, adult acinar cells are highly resistant to Kras-driven transformation and require additional factors such as Raf1 and/or Egfr to undergo malignant conversion [65].
However, not all GEMMs require inflammation to initiate pancreatic tumorigenesis. The well-established Pdx-1-Cre; LSL-KrasG12D/+;LSL-Trp53R172H/+; (KPC) model, developed by Tuveson and colleagues, revealed that concurrent mutation of TP53 not only facilitates primary tumor formation but also supports metastatic spread. These mutations are associated with dysregulation of signaling pathways implicated in pancreatic cancer progression (e.g., Erbb1/2, Egfr, Her2, Shh) and, critically, with the loss of heterozygosity (LOH) at the mutant TP53 locus. This LOH promotes chromosomal instability (CIN) and aneuploidy, in contrast to wild-type TP53 cells, which remain diploid [33].
CIN is now recognized as a hallmark of PDAC [66] and represents a frequent event in its evolution [67]. In the context of KRAS and TP53 co-mutation, CIN arises from defective DNA damage responses, disrupted mitosis, and subsequent acquisition of aneuploidy, thereby promoting tumor heterogeneity and aggressive phenotypes [32, 33, 68]. Mechanistically, TP53 loss impairs cell-cycle checkpoints and centrosome function, further propagating CIN and enabling clonal expansion of genomically unstable cells [69]. Interestingly, unlike many cancers where CIN correlates with telomere shortening [70], the KPC model preserves telomere integrity, indicating that KRAS and TP53 mutations induce CIN through telomere-independent mechanisms [68].
Genomic instability is further intensified in advanced stages, with features like tetraploidization and chromothripsis emerging, both of which are linked to enhanced cancer stemness, treatment resistance, and metastatic capacity [71-74]. Such genomic chaos fuels the development of a heterogeneous and aggressive tumor landscape, positioning CIN, driven by TP53 dysfunction and KRAS hyperactivation, as a pivotal force in PDAC pathogenesis and the potential rise of CSCs. The latter will be discussed in further detail in the following section.
Finally, other types of genetic alterations, which are now gaining attention, have been shown to play an important role in the development of pancreatic cancer and the emergence of CSCs. Splicing, a phenomenon normally performed in every cell, has attracted the attention of the pancreatic cancer field as its dysregulation could promote the expression of pro-tumoral or pro-CSC splice variants or induce an inflammatory state on its own. As described by Wan et al., the splicing factor SRSF1 could induce pancreatitis via the MAPK pathway and IL1R1 expression through mRNA stability [75]. Interestingly, p53 mutations, but not p53 KO or wild-type p53, can induce the alternative splicing of GTPase-activating proteins generating isoforms, which in turn, stimulate KRASmut activity and promote tumorigenesis [69]. Cancer stemness is, in part, a result of this alternative splicing machinery, where factors such as SF3B1 can promote characteristics such as self-renewal, proliferation or migration [76]. Apart from this, activation of certain transcription factors like HNF1A could be behind the expression of canonical stem cell factors/markers like Oct4, CD44 or EpCAM. Additionally, a transcriptional program associated to HNF1A targets was correlated with poor overall survival in patients [77]. Interestingly, Kalisz et al. have shown that HNF1A is expressed primarily in acinar cells, and its knock-out leads to tumor progression by cooperating with KDM6A and induces an sarcomatoid-like phenotype [78]. Thus, in addition to CIN and genetic chaos, alterations at the genetic level could also be key and necessary components for the tumor initiation / CSC activation cascade, although more research is still needed in this regard.
6. Genomic Instability and the Rise of CSCs in PDAC
As discussed in the previous section, genomic instability, encompassing chromosomal alterations, mutations, and structural rearrangements, generates functional and genetic diversity within PDAC tumors, and may participate in the rise of CSCs via various mechanisms discussed below [41, 67, 79, 80]. Genomic instability manifests as: (1) CIN: loss or gain of chromosomes or fragments, resulting in CSC subpopulations with diverse phenotypes. Common CIN-associated events include SMAD4 loss or GATA6 amplification [68, 81]; (2) Chromothripsis: a catastrophic chromosomal shattering and erroneous reassembly process, frequently observed in PDAC [82, 83], leading to the emergence of CSCs with increased metastatic potential or therapy resistance in other tumors such as medulloblastoma or leukemia [84, 85]; (3) Whole-Genome Duplication (WGD): a widespread feature in PDAC and other tumors that increases the likelihood of additional alterations, thus contributing to heterogeneity and aggressiveness [66, 86, 87]. The classical stepwise PanIN model proposes a gradual accumulation of mutations (KRAS → CDKN2A → TP53 → SMAD4) [88]. However, emerging evidence now supports a punctuated evolution model, where complex rearrangements, polyploidization, and chromothripsis lead to the simultaneous inactivation of multiple driver genes and rapid tumor progression [68, 89].
Beyond genetic events, tumor heterogeneity is also shaped by two conceptual frameworks: the hierarchical model, which posits that CSCs at the apex give rise to differentiated progeny [3, 90], and the clonal evolution model, in which subclonal mutations confer adaptive advantages to competing clones [91, 92]. These models are not mutually exclusive; indeed, cellular plasticity blurs their boundaries. Non-CSCs can acquire stem-like traits under stressors such as chemotherapy or radiation, thereby replenishing the CSC pool, as described above [93-95].
Genomic instability acts as a critical enabler of this plasticity. It allows transformation of normal stem cells into CSCs via accumulated mutations [96] or reprogramming of differentiated cells into CSCs, facilitated by signaling pathways such as WNT [97], NOTCH [98], and TGF-β [99]. In PDAC, where genomic instability is particularly high, these mechanisms may explain part of the dynamic and adaptive nature of CSC populations [23]. This adaptability presents a major challenge for treatment, as targeting CSCs alone may be insufficient due to continuous CSC replenishment from non-CSC populations [94, 95]. Notably, CSCs do not tolerate limitless instability. There exists a genomic instability threshold, beyond which mutations in essential housekeeping genes render cells nonviable [100]. This threshold presents a therapeutic opportunity: aneuploid cells and those with high genomic instability are more susceptible to chemotherapy [101, 102]. However, CSCs often maintain a relatively balanced instability, low enough to preserve key functions, yet flexible enough to adapt to environmental pressures [103, 104].
Thus, genetic alterations, especially KRAS and TP53 co-mutations, set the stage for chromosomal instability, a hallmark that fuels CSC plasticity and tumor heterogeneity in PDAC. Far from being static, CSCs represent a dynamic state shaped by genomic instability, contributing to treatment resistance and metastatic potential. Understanding this complexity is key to developing therapeutic strategies that not only target CSCs but also prevent their reemergence from non-stem tumor cell populations.
7. Pancreatic Cancer Stem Cells Tolerate Chromosomal Instability
Recent discoveries regarding the mechanisms that confer resistance to CSCs against stress induced by genomic instability provide valuable insight into the processes underlying their self-renewal capacity and therapeutic resistance [93, 105-108]. Stress tolerance represents a key property of these cells, including their resistance to DNA damage [109, 110], through specific mechanisms that allow survival in genetically unstable environments, including the activation of sets of signaling pathways promoting tolerance to genomic instability [93, 105, 111, 112]
As mentioned above, genomic instability is a hallmark of PDAC, promoting a high genotoxic stress environment that compromises cell viability [80, 113, 114]. This stress forces the activation of DNA repair systems as a defense mechanism, particularly in CSCs [115, 116]. CSCs aberrantly activate these repair pathways, maintaining greater repair efficiency compared to differentiated cells, which contributes to their resistance to chemotherapy [116, 117]. In pancreatic CSCs, increased expression of genes related to the cell cycle and DNA repair, such as BRCA1 (Breast Cancer type 1 susceptibility protein), has been observed in response to gemcitabine [117]. Furthermore, many sporadic or hereditary PDAC tumors present somatic or germline mutations in DNA damage response (DDR) genes, such as BRCA1/2, PALB2 (Partner and Localizer of BRCA2), and ATM (Ataxia Telangiectasia Mutated) [118]. Therefore, these genes with roles in DNA repair provide tools to tolerate genetic damage.
Among emerging mechanisms, the TGFBI (Transforming Growth Factor Beta-Induced)–ZEB1 axis has been shown to be a central modulator in CIN tolerance, initially in activated breast CSCs [119]. TGFBI, an extracellular matrix protein secreted by CSCs, induces the expression of ZEB1 (Zinc finger E-box-binding homeobox 1), a transcription factor that promotes EMT, which modulates mitotic fidelity and suppresses chromosomal errors, thereby protecting genomic stability [119]. Moreover, ZEB1 induces the antioxidant enzyme MSRB3, which limits reactive oxygen species (ROS) damage, and represses genes involved in mitotic checkpoints and damage response [119, 120]. ZEB1 also promotes stemness, invasiveness, and treatment resistance in PDAC [121-123]. Notably, knockdown of ZEB1 in highly aggressive pancreatic cancer cell lines resulted in reduced tumorigenicity in limiting dilution assays, as well as diminished tumor sphere formation and CSC surface marker expression in vitro [123], suggesting ZEB1-mediated defense against chromosomal instability in pancreatic CSCs.
On the other hand, the cGAS/STING pathway, initially recognized as an antiviral immune sensor [124], is activated in the presence of cytosolic DNA from micronuclei caused by CIN [125]. This pathway regulates multiple cellular processes, including DNA damage checkpoint signaling, repair and replication [126]. The cGAS protein acts as a decelerator of replication forks, thus reducing genomic instability [127], and its activation correlates with immune infiltration in tumors with CIN. Activation of this pathway leads to the production of IL-6, which stimulates JAK (Janus Kinase)/STAT3 (Signal Transducer and Activator of Transcription 3) signaling, promoting cell survival under genotoxic stress [126, 128]. In breast CSCs, IL-6/STAT3 promotes proliferation and immune evasion [93]. As mentioned above, in PDAC, this pathway is active in both tumor cells and their microenvironment, sometimes with greater relevance than KRAS [129, 130]. Our group has shown that the IL-6/STAT3 pathway is enriched in CSCs in pancreatic cancer [55, 130], and other studies confirm its involvement in the self-renewal and function of these cells in PDAC [50]. Since this pathway also participates in the adaptive response to DNA damage and replicative stress, its activation in pancreatic cancer CSCs could be an integral part of their defense against genomic instability.
ISG15, a gene stimulated by interferons originally linked to antiviral immunity [131], has also gained relevance in cancer due to its role in regulating replicative stress and genomic stability [132, 133]. As already mentioned, ISG15 exerts its function through ISGylation, a post-translational modification similar to ubiquitination, affecting cell cycle and replication proteins [133]. Recent studies show that ISG15 prevents replication fork collapse and protects against genotoxic damage, while its loss tonically activates Ataxia Telangiectasia and Rad3-related (ATR), increasing genomic instability [132]. As detailed above, our group has observed high ISG15 expression and elevated ISGylation levels in pancreatic CSCs, which are associated with metabolic plasticity and maintenance of the stem phenotype [43, 54]. This suggests that ISG15 is not only a functional marker of CSC in PDAC but may also be an essential mediator of their tolerance to genomic instability.
In addition, CSCs activate the ATR-Checkpoint kinase 1 (ATR-CHK1) pathway in immediate response to genotoxic agents, facilitating their survival [111]. Indeed, CSC in pancreatic cancer depend on Chk1 kinase to survive genotoxic damage induced by treatments such as gemcitabine [111]. Moreover, our group has shown that CSC in pancreatic cancer display higher telomerase activity and longer telomeres compared to differentiated tumor cells, which prevents damage at chromosomal ends and protects against genomic instability and apoptosis in CSC in pancreatic cancer [134].
Finally, many CSCs maintain a reversible quiescent state [135], which reduces their replication rate and exposure to damage, prolonging their viability even after treatments. Our group has confirmed that a subpopulation of slow-cycling CSCs in pancreatic cancer survives chemotherapy better [136], and that suppression of quiescence increases their sensitivity to agents such as gemcitabine, nab-paclitaxel, and 5-FU. Several studies describe the same subpopulations of slow-cycling stem cell-like tumor cells [16, 137, 138].
In conclusion, pancreatic CSCs integrate multiple mechanisms to tolerate genomic instability, including activation of DNA repair pathways (ATR/CHK1, BRCA1/2), telomere maintenance, control of immune-inflammatory response (cGAS/STING, IL-6/STAT3), post-translational modification (ISG15), and quiescence. These pathways enable them to resist genotoxic damage and maintain their regenerative capacity. This defensive network, though effective, also represents a therapeutic vulnerability. Interfering with these molecular nodes through combined strategies could weaken their genomic shield and enhance the effectiveness of treatments against PDAC.
8. Future Perspectives and Conclusions
The study of pancreatic CSCs has undergone a remarkable evolution over the past two decades. Initially viewed as a rare, static and hard-wired population defined by a limited set of surface markers, CSCs are now understood to represent a highly plastic, dynamic state shaped by both intrinsic genetic programs and extrinsic environmental pressures. This paradigm shift has revealed a profound complexity in CSC biology, one that reflects the broader heterogeneity of PDAC itself.
Moving forward, several key challenges must be addressed to translate this knowledge into improved patient outcomes. First, the precise identification and isolation of CSCs in both murine and human settings remains a major obstacle. The use of combinatorial surface markers and functional assays has improved our ability to define CSC subsets, yet questions persist regarding their origin, hierarchy and plasticity. Single-cell and spatial transcriptomics, combined with lineage tracing and in vivo imaging, will be essential to resolve these issues and determine whether certain CSC pools are more relevant to metastasis, therapy resistance or disease relapse.
Second, the influence of the TME, including immune cells, fibroblasts, and metabolic constraints, on CSC maintenance and evolution must be further elucidated. Emerging evidence shows that CSCs not only adapt to these external signals but also actively shape their niches, creating reciprocal interactions that reinforce malignancy. Similarly, the macroenvironmental landscape, including host factors such as inflammation and lifestyle habits (e.g., smoking, alcohol consumption, and even diabetes), is now recognized as a critical layer of CSC regulation. Integrating these “outer determinants” into experimental models will be vital for developing more prophylactic treatments and clinically relevant insights into CSC biology.
Third, the role of genomic instability, particularly CIN, alternative splicing, and structural rearrangements, in fueling CSC emergence and diversity highlights the need for targeted interventions that exploit these vulnerabilities. Therapies that push CSCs beyond their genomic instability threshold, impair their metabolic flexibility, or disrupt niche interactions may prove effective in eradicating CSCs while preventing their replenishment from non-CSC compartments.
Ultimately, the future of PDAC treatment will depend on our ability to design therapeutic strategies that reflect the dynamic, adaptive, and context-dependent nature of CSCs. A successful approach will likely require a combination of CSC-targeted agents, immunomodulatory therapies, metabolic inhibitors, and interventions aimed at altering the tumor macro- and micro-environment. As we continue to dissect the complex network of inner and outer determinants that sustain CSCs, we move closer to more precise and durable treatments for one of the deadliest forms of cancer.
Declarations
Competing Interests
The authors have declared that no competing interests exist.
Table 1 Summary of the main murine models employed in the different studies which have led to the identification of putative PDAC CSCs, including the genotype of the strain, the experimental strategy employed in the mentioned study, the primary use of the strain, main findings, novel cancer stem cell marker identified and the reference.
| Study |
Relevant Genotype |
Experimental Strategy |
Primary Use |
Key Findings |
Stem Cell Markers Identified |
| Westphalen et al. |
LSL-KrasG12D; Dclk1-CreERT; Rosa26; mTomato/mGFP(mTmG) |
In vivo lineage tracing |
Study of quiescent DCLK1-positive cells |
DCLK1-positive cells are rare, quiescent, and expand upon injury; potential adult progenitors |
DCLK1 |
| Lodestijn et al. |
Rosa26[CA]30-EYFP |
Marker-free lineage tracing |
Assess regeneration capacity |
All acinar cells contribute to regeneration post-injury; no clonal expansion with cerulein |
— |
| Jian et al. |
Dclk1-CreERT; Tff2-CreERT; LSL-Trp53+/R172H; LSL-KrasG12D |
In vivo lineage tracing |
Study of progenitor subpopulations |
TFF2-positive cells are transient amplifiers; DCLK1-positive cells contribute to homeostasis and both Generate CSCs upon transformation |
TFF2, DCLK1 |
| López-Gil and García-Silva et al. |
Pdx1-Cre; LSL-KrasG12D; Trp53+/R172H |
Cell sorting and functional assays |
Identify stem-like acinar subpopulation |
Rare acinar population with multilineage and self-renewal potential; putative CSCs with immune evasive properties which contribute to PDAC expansion |
EpCAM, SCA1, CD133, PGLYRP1 |
| Criscimanna et al. |
Pdx1-Cre; Rosa26-DTR |
Diphtheria toxin ablation |
Study of ductal cell plasticity |
Ductal cells survive ablation and contribute to regeneration; show acinar and progenitor traits |
|
| Rovira et al. |
Sox9-IRES-eGFP |
Reporter mouse line |
Ductal cell heterogeneity via organoids |
Ductal heterogeneity affects acinar-like differentiation potential |
|
| Li et al. |
Immunocompromised mice |
Xenograft assay |
Initial identification of PDAC CSCs |
CD24+/CD44+/EpCAM+ cells have highly tumorigenic potential |
CD24, CD44, EpCAM |
| Hermann et al. |
Immunocompromised mice |
Xenograft assay |
CSC chemoresistance and metastasis |
CD133+/CXCR4+ cells are highly metastatic CSCs |
CD133, CXCR4 |
| Olive et al. |
Pdx1-Cre; LSL-KrasG12D |
GEMM prenatal |
General PDAC model |
Broad transformation; lacks specificity |
|
| Hingorani et al. |
Ptf1a-Cre; LSL-KrasG12D |
GEMM prenatal |
General PDAC model |
Broad transformation; similar to Pdx1 model |
|
| Guerra et al. |
Elas-tTA; Tet-O-Cre; LSL-KrasG12Vgeo; Trp53lox/lox |
Inducible GEMM postnatal |
Acinar-specific adult PDAC model |
Tumorigenesis requires inflammation in adults |
|
Table 2 Summary of the micro- and macro-environmental stimuli or factors that can enhance the behavior or phenotypes of CSCs.
| Factor/Stimulus |
Mechanism/Pathway |
Effect on CSCs |
| Macrophages |
LL37→FPR2, P2X7R; ISG15 in response to IFNβ |
Induce and sustain CSC activity; essential for tumor formation |
| Th17 cells |
IL-17 secretion |
Promotes CSC traits |
| Fibroblasts (CAFs) |
Upregulate CD44, ALDH1, ABCG2; SPP1 expression |
Enhance CSC marker expression and stemness |
| Pancreatic stellate cells |
IL-6 secretion; Nodal/Activin pathway |
Promote CSC traits and activation |
| Low glucose/nutrient stress |
Induces OXPHOS adaptation |
Enhances stemness and therapy resistance |
| ISG15 |
ISGylation→mitochondrial renewal |
Promotes CSC metabolic plasticity; affects metformin sensitivity |
| Galactose medium culture |
OXPHPS selection |
Enriches for metabolically active, chemoresistant CSCs |
| Nicotine |
nAChRs→SHH pathway; AKT-ERK-MYC→GATA6 downregulation |
Increases stemness, EMT, dedifferentiation, metastasis |
| Cigarette smoke extract (CSE) |
CHRNA7→MAPK1→FOSL1→PAF1 |
Promotes stemness and tumor burden |
| NNK (carcinogen) |
Induces autophagy |
Promotes CSC-related chemoresistance |
| Ethanol (alcohol) |
Long-term exposure |
Enrichment of CSC-related genes and markers (in vitro) |
| New-onset type II diabetes |
Unclear |
Potential link to CSC regulation (needs further study) |
| Microbiome |
Emerging evidence |
May influence CSC dynamics and tumor progression |
References
| 1. |
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648.
[Google Scholar]
[CrossRef]
|
| 2. |
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007;67(3):1030-1037.
[Google Scholar]
[CrossRef]
|
| 3. |
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell. 2007;1(3):313-323.
[Google Scholar]
[CrossRef]
|
| 4. |
de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature. 2017;543(7647):676-680.
[Google Scholar]
[CrossRef]
|
| 5. |
Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8(5):511-524.
[Google Scholar]
[CrossRef]
|
| 6. |
Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478(7368):255-259.
[Google Scholar]
[CrossRef]
|
| 7. |
Apelqvist Å, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877-881.
[Google Scholar]
[CrossRef]
|
| 8. |
Hale MA, Kagami H, Shi L, Holland AM, Elsässer H-P, Hammer RE, et al. The homeodomain protein PDX1 is required at mid-pancreatic development for the formation of the exocrine pancreas. Dev Biol. 2005;286(1):225-237.
[Google Scholar]
[CrossRef]
|
| 9. |
Stanger BZ, Hebrok M. Control of cell identity in pancreas development and regeneration. Gastroenterology. 2013;144(6):1170-1179.
[Google Scholar]
[CrossRef]
|
| 10. |
Storz P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2017;14(5):296-304.
[Google Scholar]
[CrossRef]
|
| 11. |
Direnzo D, Hess DA, Damsz B, Hallett JE, Marshall B, Goswami C, et al. Induced Mist1 expression promotes remodeling of mouse pancreatic acinar cells. Gastroenterology. 2012;143(2):469-680.
[Google Scholar]
[CrossRef]
|
| 12. |
Johnson CL, Peat JM, Volante SN, Wang R, McLean CA, Pin CL. Activation of protein kinase Cδ leads to increased pancreatic acinar cell dedifferentiation in the absence of MIST1. J Pathol. 2012;228(3):351-365.
[Google Scholar]
[CrossRef]
|
| 13. |
Jia D, Sun Y, Konieczny SF. Mist1 regulates pancreatic acinar cell proliferation through p21 CIP1/WAF1. Gastroenterology. 2008;135(5):1687-1697.
[Google Scholar]
[CrossRef]
|
| 14. |
Pinho AV, Rooman I, Reichert M, De Medts N, Bouwens L, Rustgi AK, et al. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut. 2011;60(7):958.
[Google Scholar]
[CrossRef]
|
| 15. |
Wollny D, Zhao S, Everlien I, Lun X, Brunken J, Brüne D, et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev Cell. 2016;39(3):289-301.
[Google Scholar]
[CrossRef]
|
| 16. |
Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell. 2016;18(4):441-455.
[Google Scholar]
[CrossRef]
|
| 17. |
Lodestijn SC, van den Bosch T, Nijman LE, Moreno LF, Schlingemann S, Sheraton VM, et al. Continuous clonal labeling reveals uniform progenitor potential in the adult exocrine pancreas. Cell Stem Cell. 2021;28(11):2009-2019.e4.
[Google Scholar]
[CrossRef]
|
| 18. |
Jiang Z, Wu F, Laise P, Takayuki T, Na F, Kim W, et al. Tff2 defines transit-amplifying pancreatic acinar progenitors that lack regenerative potential and are protective against Kras-driven carcinogenesis. Cell Stem Cell. 2023;30(8):1091-109.e7.
[Google Scholar]
[CrossRef]
|
| 19. |
López-Gil JC, García-Silva S, Ruiz-Cañas L, Navarro D, Palencia-Campos A, Giráldez-Trujillo A, et al. The Peptidoglycan Recognition Protein 1 confers immune evasive properties on pancreatic cancer stem cells. Gut. 2024;gutjnl-2023-330995.
[Google Scholar]
[CrossRef]
|
| 20. |
Salas-Escabillas DJ, Hoffman MT, Brender SM, Moore JS, Wen H-J, Benitz S, et al. Tuft cells transdifferentiate to neural-like progenitor cells in the progression of pancreatic cancer. Dev Cell. 2025;60(6):837-852.e3.
[Google Scholar]
[CrossRef]
|
| 21. |
Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, et al. Duct Cells Contribute to Regeneration of Endocrine and Acinar Cells Following Pancreatic Damage in Adult Mice. Gastroenterology. 2011;141(4):1451-1462.e6.
[Google Scholar]
[CrossRef]
|
| 22. |
Fernández Á, Casamitjana J, Holguín-Horcajo A, Coolens K, Mularoni L, Guo L, et al. A Single-Cell Atlas of the Murine Pancreatic Ductal Tree Identifies Novel Cell Populations With Potential Implications in Pancreas Regeneration and Exocrine Pathogenesis. Gastroenterology. 2024;167(5):944-960.e15.
[Google Scholar]
[CrossRef]
|
| 23. |
Hermann PC, Sainz B Jr. Pancreatic cancer stem cells: A state or an entity? Semin Cancer Biol. 2018;53:223-231.
[Google Scholar]
[CrossRef]
|
| 24. |
Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep. 2019;46(6):6629-6645.
[Google Scholar]
[CrossRef]
|
| 25. |
Zhang L, Wang D, Li Y, Liu Y, Xie X, Wu Y, et al. CCL21/CCR7 Axis Contributed to CD133+ Pancreatic Cancer Stem-Like Cell Metastasis via EMT and Erk/NF-κB Pathway. PloS One. 2016;11(8):e0158529.
[Google Scholar]
[CrossRef]
|
| 26. |
Kim SK, Kim H, Lee DH, Kim TS, Kim T, Chung C, et al. Reversing the intractable nature of pancreatic cancer by selectively targeting ALDH-high, therapy-resistant cancer cells. PloS One. 2013;8(10):e78130.
[Google Scholar]
[CrossRef]
|
| 27. |
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218-2227.e5.
[Google Scholar]
[CrossRef]
|
| 28. |
Wang VM, Ferreira RMM, Almagro J, Evan T, Legrave N, Zaw Thin M, et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat Cell Biol. 2019;21(11):1425-1435.
[Google Scholar]
[CrossRef]
|
| 29. |
Fox RG, Lytle NK, Jaquish DV, Park FD, Ito T, Bajaj J, et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534(7607):407-411.
[Google Scholar]
[CrossRef]
|
| 30. |
Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014;146(1):245-256.
[Google Scholar]
[CrossRef]
|
| 31. |
Miranda-Lorenzo I, Dorado J, Lonardo E, Alcala S, Serrano AG, Clausell-Tormos J, et al. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat Methods. 2014;11(11):1161-1169.
[Google Scholar]
[CrossRef]
|
| 32. |
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457-1461.
[Google Scholar]
[CrossRef]
|
| 33. |
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469-483.
[Google Scholar]
[CrossRef]
|
| 34. |
Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63(9):2005-2009.
[Google Scholar]
|
| 35. |
Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell. 2007;11(3):291-302.
[Google Scholar]
[CrossRef]
|
| 36. |
Bailey JM, Hendley AM, Lafaro KJ, Pruski MA, Jones NC, Alsina J, et al. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene. 2016;35(32):4282-4288.
[Google Scholar]
[CrossRef]
|
| 37. |
Rajbhandari N, Hamilton M, Quintero CM, Ferguson LP, Fox R, Schürch CM, et al. Single-cell mapping identifies MSI+ cells as a common origin for diverse subtypes of pancreatic cancer. Cancer Cell. 2023;41(11):1989-2005.e9.
[Google Scholar]
[CrossRef]
|
| 38. |
Maruno T, Fukuda A, Goto N, Tsuda M, Ikuta K, Hiramatsu Y, et al. Visualization of stem cell activity in pancreatic cancer expansion by direct lineage tracing with live imaging. eLife. 2021;10:e55117.
[Google Scholar]
[CrossRef]
|
| 39. |
Burdziak C, Alonso-Curbelo D, Walle T, Reyes J, Barriga FM, Haviv D, et al. Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis. Science. 2023;380(6645):eadd5327.
[Google Scholar]
[CrossRef]
|
| 40. |
Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):441-455.
[Google Scholar]
[CrossRef]
|
| 41. |
Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646-674.
[Google Scholar]
[CrossRef]
|
| 42. |
Sainz B, Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, Cioffi M, et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015;64(12):1921-1935.
[Google Scholar]
[CrossRef]
|
| 43. |
Sainz B Jr., Martín B, Tatari M, Heeschen C, Guerra S. ISG15 Is a Critical Microenvironmental Factor for Pancreatic Cancer Stem Cells. Cancer Res. 2014;74(24):7309-7320.
[Google Scholar]
[CrossRef]
|
| 44. |
Leinenkugel G, Kong B, Raulefs S, Miller K, Roth S, Jiang H, et al. Sca-1 is a marker for cell plasticity in murine pancreatic epithelial cells and induced by IFN-beta in vitro. Pancreatology. 2022;22(2):294-303.
[Google Scholar]
[CrossRef]
|
| 45. |
Sainz B Jr., Carron E, Vallespinós M, Machado HL. Cancer Stem Cells and Macrophages: Implications in Tumor Biology and Therapeutic Strategies. Mediators Inflamm. 2016;2016:9012369.
[Google Scholar]
[CrossRef]
|
| 46. |
Zhang Y, Zoltan M, Riquelme E, Xu H, Sahin I, Castro-Pando S, et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology. 2018;155(1):210-223.e3.
[Google Scholar]
[CrossRef]
|
| 47. |
Caronni N, La Terza F, Vittoria FM, Barbiera G, Mezzanzanica L, Cuzzola V, et al. IL-1β(+) macrophages fuel pathogenic inflammation in pancreatic cancer. Nature. 2023;623(7986):415-422.
[Google Scholar]
[CrossRef]
|
| 48. |
Tu M, Klein L, Espinet E, Georgomanolis T, Wegwitz F, Li X, et al. TNF-α-producing macrophages determine subtype identity and prognosis via AP1 enhancer reprogramming in pancreatic cancer. Nat Cancer. 2021;2(11):1185-1203.
[Google Scholar]
[CrossRef]
|
| 49. |
Nallasamy P, Nimmakayala RK, Karmakar S, Leon F, Seshacharyulu P, Lakshmanan I, et al. Pancreatic Tumor Microenvironment Factor Promotes Cancer Stemness via SPP1-CD44 Axis. Gastroenterology. 2021;161(6):1998-2013.e7.
[Google Scholar]
[CrossRef]
|
| 50. |
Kesh K, Garrido VT, Dosch A, Durden B, Gupta VK, Sharma NS, et al. Stroma secreted IL6 selects for "stem-like" population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis. 2020;11(11):967.
[Google Scholar]
[CrossRef]
|
| 51. |
Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9(5):433-446.
[Google Scholar]
[CrossRef]
|
| 52. |
Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22(4):590-605.
[Google Scholar]
[CrossRef]
|
| 53. |
Nimmakayala RK, Leon F, Rachagani S, Rauth S, Nallasamy P, Marimuthu S, et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene. 2021;40(1):215-231.
[Google Scholar]
[CrossRef]
|
| 54. |
Alcalá S, Sancho P, Martinelli P, Navarro D, Pedrero C, Martín-Hijano L, et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun. 2020;11(1):2682.
[Google Scholar]
[CrossRef]
|
| 55. |
Valle S, Alcalá S, Martin-Hijano L, Cabezas-Sáinz P, Navarro D, Muñoz ER, et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat Commun. 2020;11(1):5265.
[Google Scholar]
[CrossRef]
|
| 56. |
Alcalá S, Villarino L, Ruiz-Cañas L, Couceiro JR, Martínez-Calvo M, Palencia-Campos A, et al. Targeting cancer stem cell OXPHOS with tailored ruthenium complexes as a new anti-cancer strategy. J Exp Clin Canc Res. 2024;43(1):33.
[Google Scholar]
[CrossRef]
|
| 57. |
Al-Wadei MH, Banerjee J, Al-Wadei HA, Schuller HM. Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur J Cancer. 2016;52:188-196.
[Google Scholar]
[CrossRef]
|
| 58. |
Hermann PC, Sancho P, Cañamero M, Martinelli P, Madriles F, Michl P, et al. Nicotine Promotes Initiation and Progression of KRAS-Induced Pancreatic Cancer via Gata6-Dependent Dedifferentiation of Acinar Cells in Mice. Gastroenterology. 2014;147(5):1119-1133.e4.
[Google Scholar]
[CrossRef]
|
| 59. |
Nimmakayala RK, Seshacharyulu P, Lakshmanan I, Rachagani S, Chugh S, Karmakar S, et al. Cigarette Smoke Induces Stem Cell Features of Pancreatic Cancer Cells via PAF1. Gastroenterology. 2018;155(3):892-908.e6.
[Google Scholar]
[CrossRef]
|
| 60. |
Chen X, Zhang W, Liu R, Zhu Z, Gong M, Wang Q, et al. NNK from tobacco smoking enhances pancreatic cancer cell stemness and chemoresistance by creating a β2AR-Akt feedback loop that activates autophagy. Mol Oncol. 2022;16(15):2881-2895.
[Google Scholar]
[CrossRef]
|
| 61. |
Yu W, Ma Y, Shankar S, Srivastava RK. Chronic ethanol exposure of human pancreatic normal ductal epithelial cells induces cancer stem cell phenotype through SATB2. J Cell Mol Med. 2018;22(8):3920-3928.
[Google Scholar]
[CrossRef]
|
| 62. |
Jensen MH, Cichosz SL, Hejlesen O, Henriksen SD, Drewes AM, Olesen SS. Risk of pancreatic cancer in people with new-onset diabetes: A Danish nationwide population-based cohort study. Pancreatology. 2023;23(6):642-649.
[Google Scholar]
[CrossRef]
|
| 63. |
Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178(4):795-806.e12.
[Google Scholar]
|
| 64. |
Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4(2):111-120.
[Google Scholar]
[CrossRef]
|
| 65. |
Assi M, Achouri Y, Loriot A, Dauguet N, Dahou H, Baldan J, et al. Dynamic Regulation of Expression of KRAS and Its Effectors Determines the Ability to Initiate Tumorigenesis in Pancreatic Acinar Cells. Cancer Res. 2021;81(10):2679-2689.
[Google Scholar]
[CrossRef]
|
| 66. |
Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. 2020;52(2):231-240.
[Google Scholar]
[CrossRef]
|
| 67. |
Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109-1113.
[Google Scholar]
[CrossRef]
|
| 68. |
Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538(7625):378-382.
[Google Scholar]
[CrossRef]
|
| 69. |
Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan C-H, Singh RK, et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell. 2020;38(2):198-211.e8.
[Google Scholar]
[CrossRef]
|
| 70. |
Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. 2017;18(3):175-186.
[Google Scholar]
[CrossRef]
|
| 71. |
Galofré C, Gönül Geyik Ö, Asensio E, Wangsa D, Hirsch D, Parra C, et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers. 2020;12(5).
[Google Scholar]
[CrossRef]
|
| 72. |
Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JA, Dürrbaum M, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14(17):2810-2820.
[Google Scholar]
[CrossRef]
|
| 73. |
Omabe K, Uduituma S, Igwe D, Omabe M. Deeper Insight in Metastatic Cancer Progression; Epithelial-to- Mesenchymal Transition and Genomic Instability: Implications on Treatment Resistance. Curr Mol Med. 2021;21(10):860-871.
[Google Scholar]
[CrossRef]
|
| 74. |
Liang Y, Zhong Z, Huang Y, Deng W, Cao J, Tsao G, et al. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem. 2010;285(7):4931-4940.
[Google Scholar]
[CrossRef]
|
| 75. |
Wan L, Lin KT, Rahman MA, Ishigami Y, Wang Z, Jensen MA, et al. Splicing Factor SRSF1 Promotes Pancreatitis and KRASG12D-Mediated Pancreatic Cancer. Cancer Discov. 2023;13(7):1678-1695.
[Google Scholar]
[CrossRef]
|
| 76. |
Alors-Perez E, Blázquez-Encinas R, Alcalá S, Viyuela-García C, Pedraza-Arevalo S, Herrero-Aguayo V, et al. Dysregulated splicing factor SF3B1 unveils a dual therapeutic vulnerability to target pancreatic cancer cells and cancer stem cells with an anti-splicing drug. J Exp Clin Cancer Res. 2021;40(1):382.
[Google Scholar]
[CrossRef]
|
| 77. |
Abel EV, Goto M, Magnuson B, Abraham S, Ramanathan N, Hotaling E, et al. HNF1A is a novel oncogene that regulates human pancreatic cancer stem cell properties. eLife. 2018;7:e33947.
[Google Scholar]
[CrossRef]
|
| 78. |
Kalisz M, Bernardo E, Beucher A, Maestro MA, del Pozo N, Millán I, et al. HNF1A recruits KDM6A to activate differentiated acinar cell programs that suppress pancreatic cancer. EMBO J. 2020;39(9):e102808.
[Google Scholar]
[CrossRef]
|
| 79. |
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495-501.
[Google Scholar]
[CrossRef]
|
| 80. |
Sahin IH, Lowery MA, Stadler ZK, Salo-Mullen E, Iacobuzio-Donahue CA, Kelsen DP, et al. Genomic instability in pancreatic adenocarcinoma: a new step towards precision medicine and novel therapeutic approaches. Expert Rev Gastroenterol Hepatol. 2016;10(8):893-905.
[Google Scholar]
[CrossRef]
|
| 81. |
Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578(7793):112-121.
[Google Scholar]
[CrossRef]
|
| 82. |
Li O, Li L, Sheng Y, Ke K, Wu J, Mou Y, et al. Biological characteristics of pancreatic ductal adenocarcinoma: Initiation to malignancy, intracellular to extracellular. Cancer Lett. 2023;574:216391.
[Google Scholar]
[CrossRef]
|
| 83. |
Cortés-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. 2020;52(3):331-341.
[Google Scholar]
[CrossRef]
|
| 84. |
Kats I, Simovic-Lorenz M, Schreiber HS, Sant P, Mallm JP, Körber V, et al. Spatio-temporal transcriptomics of chromothriptic SHH-medulloblastoma identifies multiple genetic clones that resist treatment and drive relapse. Nat Commun. 2024;15(1):10370.
[Google Scholar]
[CrossRef]
|
| 85. |
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27-40.
[Google Scholar]
[CrossRef]
|
| 86. |
Hu X, Zhu B, Vokes N, Fujimoto J, Rojas Alvarez FR, Heeke S, et al. The evolution of lung adenocarcinoma precursors is associated with chromosomal instability and transition from innate to adaptive immune response/evasion. Res Sq. 2024.
[Google Scholar]
[CrossRef]
|
| 87. |
Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18(7):411-424.
[Google Scholar]
[CrossRef]
|
| 88. |
Connor AA, Gallinger S. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat Rev Cancer. 2022;22(3):131-142.
[Google Scholar]
[CrossRef]
|
| 89. |
Real FX. A "catastrophic hypothesis" for pancreas cancer progression. Gastroenterology. 2003;124(7):1958-1964.
[Google Scholar]
[CrossRef]
|
| 90. |
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328-337.
[Google Scholar]
[CrossRef]
|
| 91. |
Lagasse E. Cancer stem cells with genetic instability: the best vehicle with the best engine for cancer. Gene Ther. 2008;15(2):136-q42.
[Google Scholar]
[CrossRef]
|
| 92. |
Solé RV, Rodríguez-Caso C, Deisboeck TS, Saldaña J. Cancer stem cells as the engine of unstable tumor progression. J Theor Biol. 2008;253(4):629-637.
[Google Scholar]
[CrossRef]
|
| 93. |
Baba SA, Zakeri A, Desgrosellier JS. Chromosomal instability as an architect of the cancer stemness landscape. Front Cell Dev Biol. 2024;12:1450614.
[Google Scholar]
[CrossRef]
|
| 94. |
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108(19):7950-7955.
[Google Scholar]
[CrossRef]
|
| 95. |
Bocci F, Gearhart-Serna L, Boareto M, Ribeiro M, Ben-Jacob E, Devi GR, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Natl Acad Sci U S A. 2019;116(1):148-157.
[Google Scholar]
[CrossRef]
|
| 96. |
Shiras A, Chettiar ST, Shepal V, Rajendran G, Prasad GR, Shastry P. Spontaneous transformation of human adult nontumorigenic stem cells to cancer stem cells is driven by genomic instability in a human model of glioblastoma. Stem cells (Dayton, Ohio). 2007;25(6):1478-1489.
[Google Scholar]
[CrossRef]
|
| 97. |
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843-850.
[Google Scholar]
[CrossRef]
|
| 98. |
Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97-106.
[Google Scholar]
[CrossRef]
|
| 99. |
Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415-424.
[Google Scholar]
[CrossRef]
|
| 100. |
Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101(23):8699-8704.
[Google Scholar]
[CrossRef]
|
| 101. |
Bendardaf R, Lamlum H, Ristamäki R, Algars A, Collan Y, Pyrhönen S. Response to chemotherapy (irinotecan plus 5-fluorouracil) in colorectal carcinoma can be predicted by tumour DNA content. Oncology. 2004;66(1):46-52.
[Google Scholar]
[CrossRef]
|
| 102. |
Sishc BJ, Saha J, Alves EM, Ding L, Lu H, Wang SY, et al. Defective homologous recombination and genomic instability predict increased responsiveness to carbon ion radiotherapy in pancreatic cancer. NPJ Precis Oncol. 2025;9(1):20.
[Google Scholar]
[CrossRef]
|
| 103. |
Bapat SA. Evolution of cancer stem cells. Semin Cancer Biol. 2007;17(3):204-213.
[Google Scholar]
[CrossRef]
|
| 104. |
Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3(12):895-902.
[Google Scholar]
[CrossRef]
|
| 105. |
Baba SA, Sun Q, Mugisha S, Labhsetwar S, Klemke R, Desgrosellier JS. Breast cancer stem cells tolerate chromosomal instability during tumor progression via c-Jun/AXL stress signaling. Heliyon. 2023;9(9):e20182.
[Google Scholar]
[CrossRef]
|
| 106. |
Abad E, Graifer D, Lyakhovich A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020;474:106-117.
[Google Scholar]
[CrossRef]
|
| 107. |
Skvortsov S, Debbage P, Lukas P, Skvortsova I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol. 2015;31:36-42.
[Google Scholar]
[CrossRef]
|
| 108. |
Gillespie MS, Ward CM, Davies CC. DNA Repair and Therapeutic Strategies in Cancer Stem Cells. Cancers. 2023;15(6).
[Google Scholar]
[CrossRef]
|
| 109. |
McGrail DJ, Lin CC, Dai H, Mo W, Li Y, Stephan C, et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018;23(7):2095-2106.
[Google Scholar]
[CrossRef]
|
| 110. |
Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA Damage in Stem Cells. Mol Cell. 2017;66(3):306-319.
[Google Scholar]
[CrossRef]
|
| 111. |
Venkatesha VA, Parsels LA, Parsels JD, Zhao L, Zabludoff SD, Simeone DM, et al. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia. 2012;14(6):519-525.
[Google Scholar]
[CrossRef]
|
| 112. |
Dhital B, Rodriguez-Bravo V. Mechanisms of chromosomal instability (CIN) tolerance in aggressive tumors: surviving the genomic chaos. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. Chromosome Res. 2023;31(2):15.
[Google Scholar]
[CrossRef]
|
| 113. |
Selvi S, Real CM, Gentiluomo M, Balounova K, Vokacova K, Cumova A, et al. Genomic instability, DNA damage response and telomere homeostasis in pancreatic cancer. Semin Cancer Biol. 2025;113:59-73.
[Google Scholar]
[CrossRef]
|
| 114. |
Das S, Cardin D. Targeting DNA Damage Repair Pathways in Pancreatic Adenocarcinoma. Curr Treat Options Oncol. 2020;21(8):62.
[Google Scholar]
[CrossRef]
|
| 115. |
Gronroos E, López-García C. Tolerance of Chromosomal Instability in Cancer: Mechanisms and Therapeutic Opportunities. Cancer Res. 2018;78(23):6529-6535.
[Google Scholar]
[CrossRef]
|
| 116. |
Maugeri-Saccà M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11(8):1627-1636.
[Google Scholar]
[CrossRef]
|
| 117. |
Mathews LA, Cabarcas SM, Hurt EM, Zhang X, Jaffee EM, Farrar WL. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas. 2011;40(5):730-739.
[Google Scholar]
[CrossRef]
|
| 118. |
Perkhofer L, Gout J, Roger E, Kude de Almeida F, Baptista Simões C, Wiesmüller L, et al. DNA damage repair as a target in pancreatic cancer: state-of-the-art and future perspectives. Gut. 2021;70(3):606-617.
[Google Scholar]
[CrossRef]
|
| 119. |
Sun Q, Wang Y, Officer A, Pecknold B, Lee G, Harismendy O, et al. Stem-like breast cancer cells in the activated state resist genetic stress via TGFBI-ZEB1. NPJ breast cancer. 2022;8(1):5.
[Google Scholar]
[CrossRef]
|
| 120. |
Morel AP, Ginestier C, Pommier RM, Cabaud O, Ruiz E, Wicinski J, et al. A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat Med. 2017;23(5):568-578.
[Google Scholar]
[CrossRef]
|
| 121. |
Wellner U, Brabletz T, Keck T. ZEB1 in Pancreatic Cancer. Cancers. 2010;2(3):1617-1628.
[Google Scholar]
[CrossRef]
|
| 122. |
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487-1495.
[Google Scholar]
[CrossRef]
|
| 123. |
Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19(5):518-529.
[Google Scholar]
[CrossRef]
|
| 124. |
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786-791.
[Google Scholar]
[CrossRef]
|
| 125. |
Guo X, Hintzsche H, Xu W, Ni J, Xue J, Wang X. Interplay of cGAS with micronuclei: Regulation and diseases. Mutat Res Rev Mutat Res. 2022;790:108440.
[Google Scholar]
[CrossRef]
|
| 126. |
Yan H, Lu W, Wang F. The cGAS-STING pathway: a therapeutic target in chromosomally unstable cancers. Signal Transduct Target Ther. 2023;8(1):45.
[Google Scholar]
[CrossRef]
|
| 127. |
Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv. 2020;6(42).
[Google Scholar]
[CrossRef]
|
| 128. |
Hong C, Schubert M, Tijhuis AE, Requesens M, Roorda M, van den Brink A, et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature. 2022;607(7918):366-373.
[Google Scholar]
[CrossRef]
|
| 129. |
Dosch AR, Singh S, Dai X, Mehra S, Silva IC, Bianchi A, et al. Targeting Tumor-Stromal IL6/STAT3 Signaling through IL1 Receptor Inhibition in Pancreatic Cancer. Mol Cancer Ther. 2021;20(11):2280-2290.
[Google Scholar]
[CrossRef]
|
| 130. |
Alcalá S, Martinelli P, Hermann PC, Heeschen C, Sainz B Jr. The Anthrax Toxin Receptor 1 (ANTXR1) Is Enriched in Pancreatic Cancer Stem Cells Derived from Primary Tumor Cultures. Stem Cells Int. 2019;2019:1378639.
[Google Scholar]
[CrossRef]
|
| 131. |
Munnur D, Banducci-Karp A, Sanyal S. ISG15 driven cellular responses to virus infection. Biochem Soc Trans. 2022;50(6):1837-1846.
[Google Scholar]
[CrossRef]
|
| 132. |
Wardlaw CP, Petrini JHJ. ISG15 conjugation to proteins on nascent DNA mitigates DNA replication stress. Nat Commun. 2022;13(1):5971.
[Google Scholar]
[CrossRef]
|
| 133. |
Wardlaw CP, Petrini JHJ. ISG15: A link between innate immune signaling, DNA replication, and genome stability. BioEssays: news and reviews in molecular, cellular and developmental biology. Bioessays. 2023;45(7):e2300042.
[Google Scholar]
[CrossRef]
|
| 134. |
Walter K, Rodriguez-Aznar E, Ferreira MSV, Frappart PO, Dittrich T, Tiwary K, et al. Telomerase and Pluripotency Factors Jointly Regulate Stemness in Pancreatic Cancer Stem Cells. Cancers. 2021;13(13):3145.
[Google Scholar]
[CrossRef]
|
| 135. |
Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis. 2021;12(9):835.
[Google Scholar]
[CrossRef]
|
| 136. |
Cioffi M, Trabulo SM, Sanchez-Ripoll Y, Miranda-Lorenzo I, Lonardo E, Dorado J, et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut. 2015;64(12):1936-1948.
[Google Scholar]
[CrossRef]
|
| 137. |
Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26(7):611-623.
[Google Scholar]
[CrossRef]
|
| 138. |
Ambrosini G, Dalla Pozza E, Fanelli G, Di Carlo C, Vettori A, Cannino G, et al. Progressively De-Differentiated Pancreatic Cancer Cells Shift from Glycolysis to Oxidative Metabolism and Gain a Quiescent Stem State. Cells. 2020;9(7):1572.
[Google Scholar]
[CrossRef]
|


 ,
Diego Navarro
1,2,4
,
Diego Navarro
1,2,4