1. Introduction
Glioblastoma (GBM) remains one of the most aggressive and therapy-resistant primary brain tumors. GBM accounts for approximately 50% of the brain and CNS malignant tumors in the United States [1]. Despite significant advances in cancer treatment modalities, the prognosis for GBM remains dismal. GBM has been recognized for its extensive interpatient as well as intratumoral heterogeneity. This is in part contributed by the fact that GBM doesn’t have a single oncogenic driver and can be a result of multiple different mutations. The intratumoral heterogeneity arises from the presence of varied cell-types, including but not limited to, tumor cells, immune cells, glial cells, neuronal cells and stem-like cancer cells [2].
John Dick’s group first identified the presence of tumor initiating cells in acute myeloid leukemia. They isolated a subpopulation of CD34+, CD38- cells that resulted in large colony forming progenitors in SCID mice and were less mature than colony forming cells [3]. The group also identified the high self-renewal potential of this population of cells by serial transplantation. They further identified that these cells could differentiate and recapitulate the donor’s disease in mice, both contributing to the stem-like characteristics of these tumor initiating cells [4]. Less than a decade later, cancer stem cells (CSCs) were first identified in solid tumors. Michael Clarke’s group identified a small population of CD44+/CD24- that generated tumors with a diverse cell population as well as a population of CD44+ cells [5]. Eventually, a plethora of cancer stem cells were identified in different solid tumors with a variety of cell surface markers, including GBM [6].
2. Cancer Stem Cells in Glioblastoma
The first stem-like cancer cell in tumors involving the CNS was identified as a clonogenic population of neural stem-like cells from human cortical glial tumors that withstood anchorage and serum withdrawal and expressed astroglial and neuronal markers while also showing similarities with normal brain derived clones [7]. Soon after, CSCs were isolated from brain tumors by enriching for CD133+ cells. These were reported to show long term self-renewal as well as the potential to differentiate [8]. Similar observations were made in pediatric brain tumors [9]. Shortly after, multipotent CSCs in GBM with long-term self-renewal potential were identified. They also successfully demonstrated that these CSCs withstood serial transplantations giving rise to tumors in vivo [10]. The Dirks lab then went on to show that tumors arising from patient-derived CD133+ xenografts resembled patient tumors [11].
GBM CSCs have been identified for their similarity with neural stem or progenitor cells. These CSCs express a variety of transcription factors as well as cell surface markers that coincide with normal neural stem cells, including CD133, SSEA-1, Nestin, SOX2, L1CAM, and many more. All genes introduced throughout this review can be found in Table 1 with their corresponding references. These markers are useful for identifying CSCs but also carry a risk of false positives since most tissues house various stem cells that show varied expression of these markers [12]. Other ways to isolate these CSC populations include enrichment in serum-free neurosphere cultures and can be further validated by limiting dilution assays.
Multiple groups have performed single cell RNA sequencing on primary patient-derived GBM samples. Despite using different language to describe the cell types within these samples, all groups found that the tumor cells were heterogeneous in nature and demonstrated dynamic transcriptional changes. Suva and Tirosh characterized GBM into four main subsets: AC-like, MES-like, OPC-like, or NPC-like [13]. Similarly, other groups found that GBM cells exist along an axis that differentiates between proneural and mesenchymal [14]; an axis that includes progenitor and differentiated cells [15]; or an axis defined as a neurodevelopment-injury axis [16]. The cell states defined by Suva and Tirosh are associated with distinct mutations. For example, cells with an OPC-like state often have amplifications of the PDGFRA gene while cells with an AC-like state have amplifications of EGFR. In GBM, these mutations are mutually exclusive, meaning each cell can only have one of these mutations. However, subpopulations of OPC-like and AC-like cells can occur within the same tumor. Of note is that these transcriptional changes between cell states in GBM samples are non-static. Cells with an NPC-like transcriptional signature which are implanted into immunocompromised mice can give rise to MES-like cells in the growing tumor [13]. There is evidence of cells that exhibit hybrid states, such as AC-like/MES-like and AC/OPC-like. These may indicate a possible plasticity exhibited by CSCs including the ability to transition between different states [13,17]. This, in part, would also contribute to the observed intra-tumoral heterogeneity in GBM.
3. Therapy Resistance in GBM
Current standard of care therapy for the treatment of GBM involves surgical resection combined with radiation and chemotherapy using the alkylating agent, temozolomide (TMZ) [18]. Ionizing radiation induces DNA damage in the form of double strand breaks. These breaks can initiate DNA repair mechanisms that rectify the breaks [19]. Pre-clinical evaluation of CD133+ CSCs showed that CSCs had more robust and efficient repair systems which allowed them to thrive better than regular tumor cells [20]. Furthermore, this was observed clinically, wherein accumulation of CD133+ cells were observed in recurrent primary GBM as well as after treatment with high dose radiation [21]. Various other genes have been reported to be upregulated in specific GBM CSCs that tend to protect cells from DNA damage and confer radioresistance. These include but are not limited to RAD51, ATM, BRCA1 and BRCA2 [22,23].
One cannot talk about therapeutic resistance in GBM without bringing up MGMT, one of the most well-studied repair proteins in the field. MGMT promoter methylation has been clinically associated with better response to TMZ. Studies have found that CD133+ cell populations in GBM tend to express MGMT which confers resistance to TMZ [24,25]. Apart from this, evidence of the presence of ABC transporters like ABCG1 and ABCB2 and their overexpression in CSCs tend to provide resistance to TMZ [26,27]. In the presence of an ABC transporter inhibitor, GBM CSCs tend to become sensitive to these treatments [27]. Other proposed mechanisms of action to TMZ resistance include p53 mediated cell cycle arrest and senescence while also showing that p53-deficient cells were more resistant to TMZ [28]. Sundar et al. successfully showed that, in their model, upon treatment with standard-of-care and targeted therapies, stem cells did not demonstrate cell death whereas there was an increase in the number of cells undergoing senescence [29]. It has also been shown that treatment with TMZ results in upregulation of stemness markers like SOX2 and OLIG2 which may point towards potential plasticity of CSCs [30].
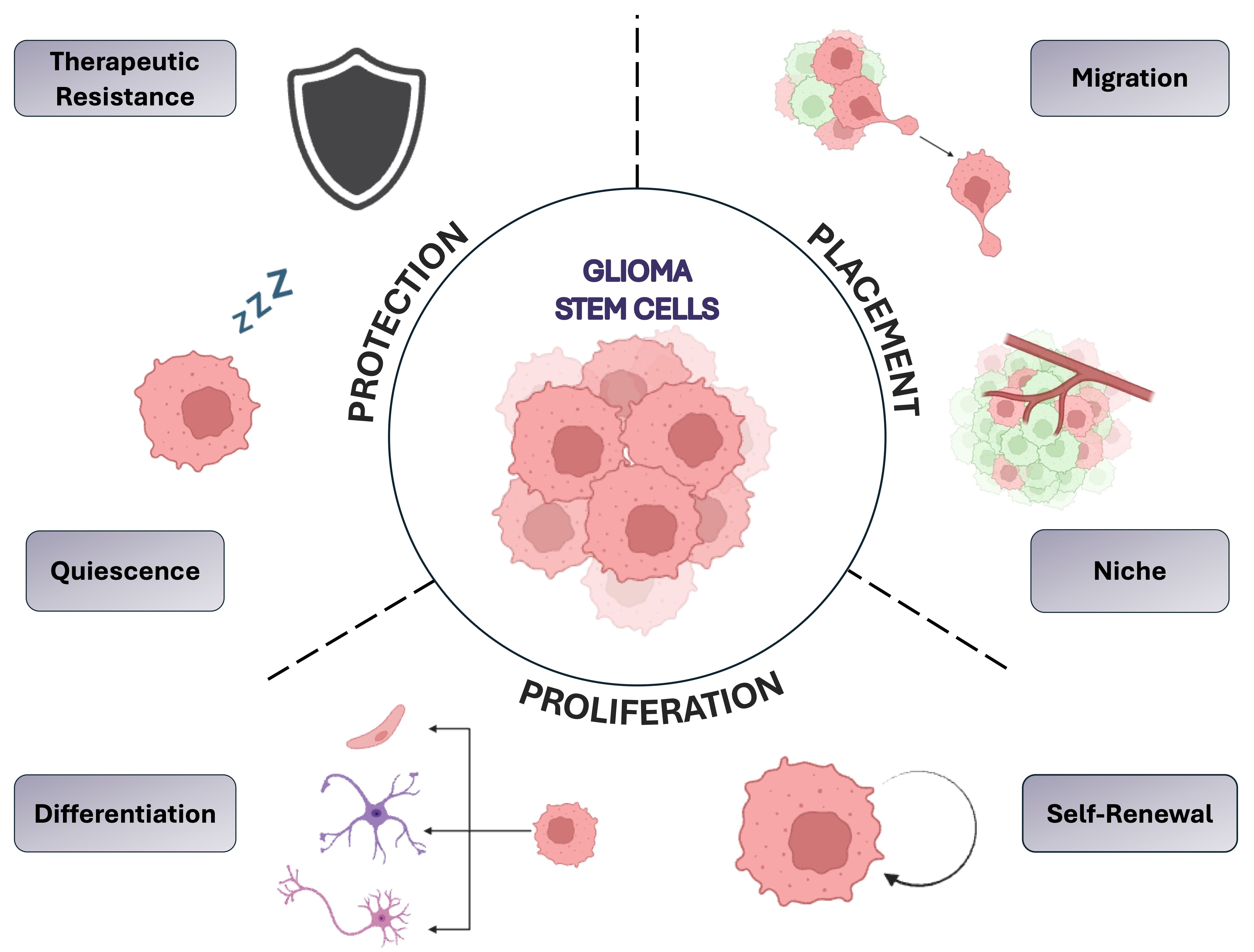
The Burma lab showed that ionizing radiation triggers senescence and senescence associated secretory phenotype (SASP) in the brain. GBM cells tend to be more aggressive in this SASP environment. The group also found that this senescence and tumor aggressiveness is p21 dependent and demonstrated that senescence was rescued in a p21 knockout mouse [31]. Figure 1 illustrates three groups of properties which drive heterogeneity and plasticity in GBM, including protection, placement, and proliferation.
4. Cell Cycle Plasticity and Quiescence in CSCs
The single cell sequencing data showed cycling cells and non-cycling cells and established hybrid states within GBM as mentioned above. Quiescence is one of the key components that provide CSCs their plasticity and the ability to “toggle” within the cell cycle. Activation of cells from the quiescent state is shown to be a key factor responsible for GBM progression [32]. The Dirks Lab showed that CSCs are slow cycling. They serially transplanted xenografts and sequenced the resulting tumors. They found that the CSC population was consistent with previous studies as being quiescent or cell cycling and the fast proliferating or actively cycling cells had insubstantial self-renewal [33]. Furthermore, it was proved that CSCs, even in culture, can be highly proliferative. Sustained inhibition of kinase signaling or, in other words, in the presence of targeted therapy, the culture is enriched for slow cycling Ki67-negative CSCs. They further identified that not only the slow-cycling cells were enriched in primitive neurodevelopmental signatures but also found various Notch pathway genes being upregulated which was further validated by the identification of Notch-positive cells which were depleted in proliferation markers in primary GBM tumors. This switch seemed to be accompanied by dynamic histone acetylation and demethylation [34]. It was also observed that a subpopulation of slow-cycling cells in GBM could recapitulate tumors in vivo. This population was identified as a set of dye-retaining cells [35]. These cells were further characterized and distinguished from fast-cycling cells based on their metabolic profiles. Slow-cycling cells relied on mitochondrial oxidative phosphorylation and displayed increased invasion and therapeutic resistance whereas the fast-cycling cells preferred the anaerobic glycolytic pathway [36]. This pivotal study highlighted that intratumoral cellular heterogeneity goes in hand with metabolic diversity.
Furthermore, a set of conserved gene signatures of quiescent cancer stem cells in murine models and compared it to pre-existing expression data from human single-cell clusters was established. They identified a 118-gene signature in quiescent cells that remain conserved in primary and recurrent GBM. They also identified a cell surface receptor, F3, which is a part of this gene set and was used to identify quiescent cells. The cells positive for F3 also showed enhanced self-renewal and tumor initiation potential, even with serial transplantation [32]. F3 has also been found to be a critical regulator for therapeutic resistance and oncogenic senescence in GBM, making it a potential therapeutic target [37].
5. Cross-Talk Between CSCs and Non-Tumor Cells
Although we’ve been successful in understanding cells’ transition in and out of a quiescent state, it is important for us to understand that there are a plethora of factors at play that work in symphony to dictate cell fate. For example, Jeremy Rich’s group established that there is abundant crosstalk between differentiated tumor cells and GBM CSCs. The group found that the IL6 receptors, IL6Rα, and gp130, the ligand that can be secreted by many different types of cells including CSCs, is essential for GBM CSC maintenance and proliferation [38]. This activates downstream STAT3 signaling which is crucial in stem cell state as well [39]. In the presence of differentiated tumor cells, CSCs thrive and enhance tumor growth through multiple mechanisms. Differentiated glioma cells express the ligand BDNF while the GBM CSCs express the cognate BDNF receptor, while also uncovering the role of BDNF signaling in tumor growth via VGF expression. These results highlight the crosstalk between these differentiated tumor cells and GBM CSCs as well as CSCs themselves [40]. Furthermore, the group also identified that these CSCs produce the bone morphogenic proteins (BMP) antagonist, Gremlin-1, against endogenous BMP, preventing their differentiation into the astrocytic lineage and maintaining the intratumoral hierarchy [41].
Another study done in 2022 found an interesting paradigm of interactions between transformed neural stem cells and wild-type neural stem cells isolated from mice subventricular zone. The group co-cultured these cells and observed that the growth rate of the wild-type neural stem cells reduced substantially when grown in the presence of transformed neural stem cells. This was also confirmed by observing lowered levels of phospho-histone H3. Transcriptional profiling revealed that the co-culture resulted in the activation quiescence and required cell-to-cell contact to do so [42].
The crosstalk does not just exist between tumor cells. Frank Winkler’s group created a novel method for the selection of network-connected glioma cells. The group had previously demonstrated tumor microtubule (TM) driven connections that allow gap junction permeable exchange of small molecules like that in astrocytes. They used high-resolution microscopy to detect TM-connected cells. They also found that TM connectivity is associated with high tumorigenicity and poor prognosis. Furthermore, these TM-positive glioma cells tend to have distinct expression profiles associated with high stem cell markers in humans [43]. This group also showed the existence of calcium ion communication in these glioma networks that eventually activate NFkB signaling within the network of cells [44]. Tumor microtubes have also been shown to aid in the transfer of mitochondria from the astrocytes to the tumor cells in a GAP43-dependent manner. This allows for tumor cells to have increased metabolic activity and promotes self-renewal and tumorigenicity [45].
The extent of these interactions does not end here. Based on recent studies, when GFP-positive GBM CSCs were grown in cortical organoids, a subpopulation of the non-malignant cells not only carried the GFP transcripts but expressed low but detectable levels of GFP [46,47]. Another study found that CSCs in the inherently hypoxic pseudopalisading regions of GBM tumors produce specific chemokines that recruit early myeloid-derived suppressor cells. These cells in turn produce growth factors like FGF11 which may promote the growth of GBM via the FGFR1 receptor [48]. Moreover, the Rich lab found that tumors arising from CSCs, not only produced more vascular and hemorrhagic tumors but also proved that these CSCs were a source of VEGF that promoted endothelial cell migration and vessel formation [49]. This indicates that GBM CSCs not only interact with various cells within the tumor community but are also involved in a transactional relationship with many of them. The ability of CSCs to engage in varied interactions and transitions between different states significantly contributes to the diversity of niches within GBM.
6. Niches Within Glioblastoma Tumors
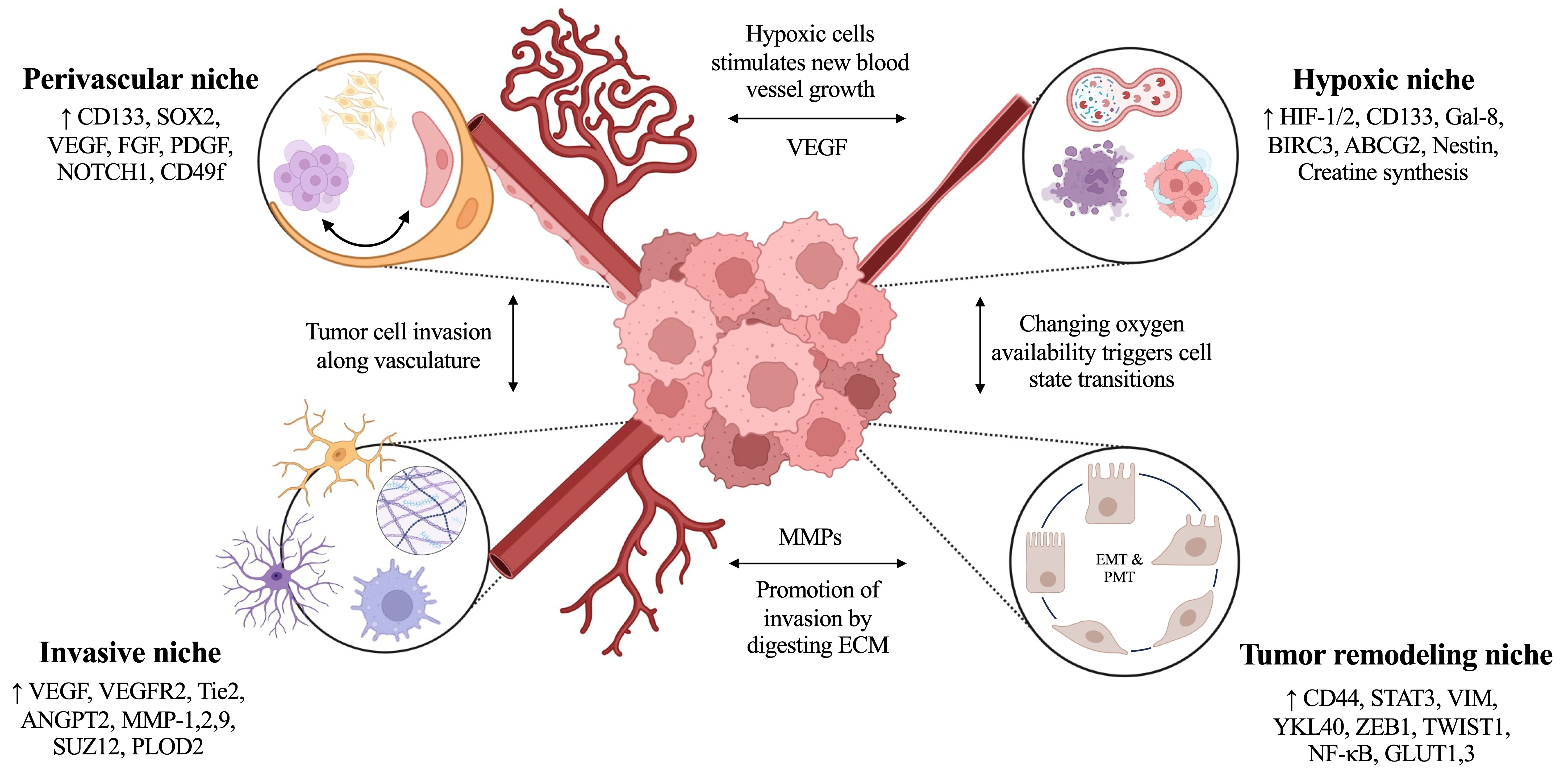
Multiple spatially distinct niches have been proposed in GBM tumors, including hypoxic, perivascular, invasive, and immunosuppressive niches [50,51]. Of these, the most is known about perivascular and hypoxic tumor zones which we review below. Figure 2 outlines distinguishing markers expressed and associated crosstalk between tumor niches.
6.1 The hypoxic, pseudopalisading niche
When vasculature, and therefore oxygen delivery, is disrupted by thrombosis or other blockage of blood flow, this creates necrosis. Nearby cells in the perinecrotic area migrate away from the disruption and towards more permissive environments. This creates a wave of increased cell density around the necrotic area, a pathologic structure termed a pseudopalisade. Upon further examination, cells within pseudopalisading regions were found to be less proliferative due to their exposure to hypoxia within the tumor, but also more apoptotic and motile as a response to this environment. Pseudopalisading around necrosis, and its opposite, excessive microvascular growth, are both distinguishing histopathological hallmarks of GBM tumors versus other, less aggressive forms of astrocytomas [52].
Tumor cells in the hypoxic niche also express biomarkers associated with poor survival, tumor cell migration and invasion, therapeutic resistance, immunosuppression, proneural-mesenchymal transition (PMT) and overall disease aggression [53]. The transcription factor hypoxia inducible factor 1 (HIF-1) is one of the central drivers of these characteristics [54]. HIF-1 is comprised of two subunits, α and β, which work together to heterodimerize and localize in the nucleus to control gene expression [55]. When HIF-1 is active and able to reach the nucleus, due to low microenvironmental oxygen, HIF-1 induces transcription of vascular endothelial growth factor (VEGF) by hypoxic GBM cells, which stimulates local angiogenesis, thus relieving the hypoxia and expanding the vascular niche area [49]. However, the process of angiogenesis is dysregulated in GBM, causing the growth of leaking, fenestrated vessels which are susceptible to collapsing. Vessel collapse promotes further hypoxia and necrosis throughout the tumor. Hypoxia is also known to drive CSC proliferation and clonogenicity in both GBM patient tumors and neurosphere cultures, particularly in cells expressing the stemness marker CD133 due to their ability to modulate the MAPK and PI3K/AKT signaling pathways [56,57]. A gain of function study linked the oxygen-stable form of HIF-1α to upregulated CD133 expression and several-fold increases in clonogenicity in GBM [56], concluding that HIF-1α is largely responsible for these hypoxic effects. As CSCs and CD133 expression are significant contributors to therapeutic resistance and tumor recurrence in GBM, the hypoxic niche drives the hallmark aggression of this disease [21].
Autophagy, a process of recycling cellular components to maintain homeostasis via lysosomes, has been found to play an important role in CSC maintenance [58]. In GBM, the process of autophagy is dysregulated in a way that promotes further therapeutic resistance and immunosuppression within the tumor. Through analysis of publicly available databases, such as The Cancer Genome Atlas (TCGA), GBM patient samples with high expression of HIF-1α were found to correlate with upregulated stemness and autophagy. Galectin-8 (Gal-8), known as a poor prognostic indicator, has been identified as a major facilitator of hypoxia-induced autophagy by fueling CSC maintenance in the hypoxic niche [58]. Here, Gal-8 was found to bind to the Rag-Ragulator complex on the surface of lysosomes, preventing the mechanistic target of rapamycin complex 1 (mTORC1) from binding and ultimately inactivating it. Normally, mTORC1 must phosphorylate the transcription factor EB (TFEB) to block nuclear translocation. However, when mTORC1 is inactive and unable to perform this phosphorylation, TFEB can enter the nucleus and activate genes associated with autophagy, such as autophagy proteins 5 and 7 (ATG5,7). Since mTORC1 is responsible for maintaining anabolic and catabolic homeostasis [59], while TFEB encourages autophagy, the Gal-8-mTOR-TFEB signaling axis was found to increase both autophagy and stemness preservation [58].
Recently, an inhibitor of apoptosis protein (IAP), called baculoviral IAP repeat containing 3 (BIRC3), has been linked to increased survival adaptation and therapeutic resistance in GBM tumors. BIRC3 inhibits apoptosis by influencing E3 ligases and signaling pathways such as the TNF, NF-κB, STAT3, and PI3K pathways, fueling resistance to temozolomide (TMZ) and radiotherapy (RT) in both GBM patient samples and cell lines [60]. CSCs which overexpress BIRC3 were also found to stimulate CD133 and ATP-binding cassette super-family G member 2 (ABCG2) expression, which are both stem cell markers, while BIRC3 knockdown reversed this effect [61]. Another stemness marker, Nestin, was also evaluated and increased BIRC3 expression was found to correlate with higher Nestin expression, further implicating BIRC3 with stemness regulation. Bone morphogenic protein 4 (BMP4), known for its role in central nervous system (CNS) development during embryogenesis, has been found to promote CSC differentiation towards astroglial cell types in GBM resulting in improved survival in mouse xenografts [62]. Data from the IVY Glioblastoma Atlas Project (IVY GAP) database revealed an interesting dynamic between BIRC3 and BMP4 within the distinct niches of GBM tumors, where the hypoxic niche demonstrates high BIRC3 expression with low BMP4 expression, while the opposite is true of the perivascular niche. Further analysis showed that the influence which BIRC3 has over CSCs is dependent on BMP4 signaling inactivation, and remarkably, BMP4 inactivation can restore stemness in GBM cells with BIRC3 knockdown [61]. These in vitro findings were also upheld in vivo studies, as high BIRC3 expression in mouse xenografts resulted in increased tumorigenicity, but xenografts established with BIRC3 knockdown cells had improved survival and reduced tumor burden. The hypoxic niche of GBM tumors is recognized as a source of CSC maintenance and reprogramming; although further studies are required to fully characterize the roles of BIRC3 and BMP4 in GBM, these proteins may be paramount for future therapeutics targeting the hypoxic niche and CSCs in GBM.
GBM tumors are described as immunologically ‘cold’ due to their notably small population of tumor-infiltrating lymphocytes (TILs). This contributes to their immunosuppressive nature, along with the presence of microglia, tumor-associated macrophages (TAMs), regulatory T-cells (Tregs), and myeloid-derived suppressor cells (MDSCs) within their TME [63-65]. New literature revealed that creatine metabolism may be an overlooked signature in tumor-associated myeloid cells (TAMCs) within GBM tumors. Normally, creatine is synthesized in the kidneys; however, in the hypoxic niche of several patient samples, de novo creatine synthesis was identified as a way for TAMCs to nourish the tumor, acting as “feeder-cells” [66]. This is in contrast to previous reports that state GBM samples contain less creatine than normal brain tissue. However, decreased creatine is also characteristic of necrosis which can occur throughout the tumor [67]. Therefore, low creatine throughout the tumor may indicate the presence of necrotic tissue within bulk tumor samples, further contributing to the heterogeneity of GBM. Creatine synthesis is limited by the enzymes glycine amidinotransferase, mitochondrial (GATM) and guanidinoacetate N-methyltransferase (GAMT), where GATM was found to be heavily influenced by hypoxia and acidity [66,68]. HIF-1α was found to be the central driver of this dynamic, regulating de novo creatine synthesis and absorption. This creatine phenomenon was exclusively investigated in the hypoxic, pseudopalisading niche of GBM tumors, finding that the hypoxic niche is reliant on creatine uptake as a self-preservation effort against metabolic stress. Extreme limiting dilution assays (ELDAs) indicated a notable increase in clonogenicity in GBM CSCs and patient-derived xenograft cells following treatment with creatine [66]. TAMCs were also found to break down arginine to synthesize ornithine and putrescine, two polyamines of basic pH, which help neutralize the acidity within the TME [69]. By synthesizing these polyamines, TAMCs effectively provide a path toward survival for other cells in the tumor, contributing to its overall immunosuppression. Additional research is required to elucidate the entire role of creatine, polyamines, and TAMCs in GBM tumors, however, it is clear that each of these components further promote the aggressive yet elusive nature of the hypoxic niche.
6.2 The perivascular niche
GBM tumors are highly vascularized and CSCs use these aberrant vessels as paths for invasion [66,70]. While often leaky and tortuous in nature, these vessels serve to deliver oxygen and nutrients to the tumor, aiding tumor cell proliferation. The perivascular niche (PVN) of GBM tumors has several other identifying features, including the presence of CSCs expressing markers such as CD133, CD49f, and SOX2, neural stem cells (NSCs), and presence of VEGF, which may be derived from gene expression in hypoxic niche tumor cells [49,50,71,72]. Along with VEGF, other growth factors contribute to the hallmark malformed vasculature, including fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) [50,73]. Transdifferentiation between GBM cells and endothelial cells is a phenomenon which may be controversial to some, however, it is another factor that stimulates further vessel recruitment and development [70]. Unlike healthy vasculature, the vessels which infiltrate GBM tumors lack critical structural components, including the basement membrane of the vessels, and endothelial cells behave abnormally which increases potential leakage out of the vascular space and into the locally surrounding niche.
Immunohistochemical staining of GBM patient samples also indicated that the NOTCH1 pathway is active in cells of the PVN, which is known to support stemness of CSCs. NOTCH1 activation is also known to occur in endothelial cells which reside along GBM cells, further promoting stem cell maintenance in the PVN. Although the surplus of vasculature and excessive signalling alludes to an invasive, motile phenotype in the PVN, these cells were actually found to be slow-cycling and relatively stagnant, including along the invasive front of the tumor [71]. However, cells in the PVN do behave similarly to reactive astrocytes such that they react to nearby injury from surgical trauma, which may also be referred to as a ‘malignant healing response.’ Knockdown of NOTCH1 in nude mice resulted in a decreased overall population of cells in the PVN and decreased vessel co-option, highlighting the influence of the NOTCH1 signaling pathway within the PVN [71].
The PVN of GBM tumors contains vessels which can form in many different ways, including (i) angiogenesis, (ii) vasculogenesis, (iii) vascular co-option, (iv) vascular mimicry, and (v) transdifferentiation between GBM and endothelial cells [74]. Angiogenesis is defined as the process of forming new vasculature from previously existing vessels, and this appears to be the tumor’s preferred method of vessel development [75]. Previously mentioned growth factors, such as VEGF, are fundamental components which drive angiogenesis and have become therapeutic targets in GBM, however targeting these processes remains a challenge. Bevacizumab, also known by the brand name Avastin, is a therapeutic targeting angiogenesis by way of VEGF-binding. Despite showing promising results in preclinical studies, clinical studies show that Bevacizumab unfortunately cannot overcome the aggressive nature of GBM tumors, providing minimal overall therapeutic benefit [75,76].
Vasculogenesis is another mechanism used to form new blood vessels in a tumor, where bone marrow-derived endothelial progenitor cells and tumor-associated macrophages are enlisted for vessel formation; this tends to occur during more advanced stages of tumor development [74]. In GBM, vessels can also take part in vessel co-option which is characterized by tumor cells being hijacked in a manner that promotes migration toward invasive cells, giving rise to the histopathologic phenomenon called perivascular satellitosis [75,77]. Histopathology describes perivascular satellitosis as a distinct grouping of tumor cells invading into the Virchow–Robin spaces, which are fluid-filled areas that surround nearby vasculature [78]. Recently, vessel co-option has been linked to cellular reprogramming in GBM, where one study identified a connection between vessel co-option and an invasive, therapy-resistant, slow-cycling phenotype [77]. Here, transcriptomic analysis of patient samples suggested that treatment with chemotherapy induced a cell state which is consistent with features of the mesenchymal subtype of GBM, where these cells obtained astrocyte-like traits and an increased ability to repair DNA damage caused by therapeutics. To track and isolate these cells– referred to as the VC-resist (vessel co-opting and resistant) cells– the authors sorted GBM cells by high and low expression of Nestin, where VC-resist cells express high levels of Nestin. Interestingly, upon sorting and segregating these Nestin-high, VC-resist cells resulted in a gradual transition back to a Nestin-low phenotype, indicating that this cell state is not fixed, but rather partially reversible [77].
6.3 The invasive niche
While both the hypoxic and perivascular niches of GBM tumors support the overall invasiveness of the disease, the tumor cells invading through the normal brain structure also find themselves in a new and separate niche which we call the invasive niche. As gradients develop throughout the tumor, forming the hypoxic and perivascular niches, single GBM cells or cell clusters are induced to invade surrounding healthy tissue [50]. Individual tumor cells migrate along the basement membrane of vasculature, as well as through the white matter tracts of the brain [50,79]. Recent literature reports a “normal-to-tumor transition” of vasculature within the invasive niche of GBM tumors, where vascular heterogeneity and remodeling is largely mediated by VEGF and vascular endothelial growth factor receptor 2 (VEGFR2) [80]. In healthy brain tissue, the VEGFR2 pathway signals for angiogenesis to establish vasculature– including that of the blood-brain barrier (BBB)– along with the tunica interna endothelial cell kinase 2 pathway (Tie2) [80]. Within the Tie2 pathway, angiopoietin-2 (ANGPT2) serves as an antagonist and high expression of both VEGF and ANGPT2 are associated with worse clinical outcomes in GBM [80,81]. The reported vascular transition is directly linked to spatial location within the tumor, with ANGPT2/Tie2 signaling mediating changes in the core of GBM tumors, while VEGF/VEGFR regulates this transition around the peripheral edges of the tumor [80]. Vasculature in the invasive niche often appears enlarged, exploiting increased permeability in order to support invasion and immunosuppression throughout the tumor [50,80]. Using a GBM mouse model with the EGFRVIII mutation and silenced p53/PTEN to model spontaneous GBM development, time-lapse imaging of the invasive niche revealed that this vascular transition throughout the tumor is initiated by a vasodilation-vasoconstriction dynamic occurring within the invasive niche. When treated with a novel anti-Tie2 antibody, vessel abnormalities were corrected in the EGFRVIII mouse models, suggesting that Tie2 may assist in regulating this normal-to-tumor transition within GBM vasculature [80].
Matrix metalloproteases (MMPs) are another critical component in GBM invasion, which are part of the zinc-dependent proteolytic endopeptidases family [82]. MMPs degrade components of the extracellular matrix (ECM) of GBM tumors, including collagen, fibronectin, and proteoglycans, allowing for ECM remodeling [82,83]. MMPs appear to have heterogeneous effects on GBM tumors, where an extensive bioinformatic analysis of MMPs in GBM revealed that different MMPs are expressed in different tumor phenotypes, leading to varying survival outcomes. This study states that MMPs 1, 2, 7, 9, 11, 14-16, and 26 were expressed significantly more in GBM samples as compared to healthy brain tissue, where five of these nine MMPs are regulated by the transcription factor Suppressor of Zeste homolog 12 (SUZ12) [82]. Another study investigating the effects of microRNAs (miRNAs) on GBM tumorigenesis reported a mutually antagonistic dynamic between miRNA-105 and SUZ12 [84]. Interestingly, high expression of miRNA-105 halted proliferation and invasion of GBM, while overexpression of SUZ12 reinstates these effects [84]. Therefore, by regulating the expression of SUZ12, the invasive nature of GBM may be able to be controlled through its connection to each of these critical MMPs. Additionally, miRNA-105 may present great therapeutic potential for high-grade gliomas [84]. It is important to note that while each of the previously listed MMPs certainly contribute to GBM invasion, MMPs 1, 2, and 9 may propel much of this effect. Ample evidence has been reported indicating that high expression of MMPs 1, 2, and 9 serve as indicators of poor survival outcomes [83]. In general, silencing MMPs in GBM improves prognosis and suppresses the invasive tendency of this malignancy.
Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 (PLOD2) also supplements GBM invasion, where an extensive study found consistent PLOD2 overexpression in GBM across 800+ samples from seven different databases [54]. As its name suggests, PLOD2 facilitates collagen crosslinking which is directly correlated to GBM’s ability to invade. When PLOD2 is overexpressed in patient tumors, crosslinking of collagen within the ECM is augmented such that ECM stiffness increases, making it easier for tumor cells to invade and metastasize [54]. Notably, HIF-1α was found to regulate PLOD2, further supporting the idea that the hypoxia throughout GBM tumors significantly promotes invasion [54]. As PLOD2 is so closely connected to GBM invasion, overexpression is also indicative of increased tumor grade and worse survival outcomes. Additionally, PLOD2 contributes to invasion and metastasis in other solid tumors, including melanoma and prostate cancer [54,85]. Although PLOD2 inhibitors such as Minoxidil are available, little research has been completed to investigate the effects in GBM; therefore, PLOD2 may serve as a viable therapeutic target for GBM both as a monotherapy and within combination therapies.
6.4 Tumor remodeling as a niche
GBM tumors are highly adaptive, constantly evolving with disease progression and in response to therapeutics. In order to overcome cellular barriers, GBM tumors can remodel tumor tissue, surrounding healthy tissue, and its metabolism in order to thrive. While transitions such as epithelial-to-mesenchymal transition (EMT), or the more brain tumor-specific term proneural-to-mesenchymal transition (PMT), are not traditionally referred to as forms of tumor remodeling, these transitions can be weaponized by the tumor for self-preservation purposes.
PMT describes a transition from the proneural to the mesenchymal GBM tumor subtypes, where the mesenchymal subtype is more aggressive and invasive than the proneural subtype, creating an advantageous opportunity for the tumor to proliferate and invade. This transition can occur upon treatment with chemotherapy or radiotherapy, fueling therapeutic resistance [86]. Additionally, PMT is observed upon GBM recurrence, eliciting overexpression of CD44, STAT3, VIM, and YKL40 with decreased OLIG2 and PTEN [86]. Dynamics within the hypoxic niche can also induce transitions towards the mesenchymal subtype, where HIF-1α takes part in a signaling axis with zinc finger E-box-binding homeobox 1 (ZEB1), where ZEB1 also contributes to stem cell maintenance [86,87]. Twist-related protein 1 (TWIST1) is a transcription factor expressed by mesenchymal GBM subtypes, which is also controlled by HIF-1α, further underscoring the destructive power of HIF signaling and the hypoxic niche [86]. TWIST1 is closely connected to EMT and unfavorable patient outcomes, enhancing invasive capabilities of GBM tumors throughout ex vivo and in vivo studies by remodeling the cytoskeleton of tumor cells and interfering with cell-substrate interactions [86]. Identifying biomarkers which are present in both proneural and mesenchymal subtypes may provide valuable insight as to how tumor remodeling is initiated. One marker that these subtypes have in common is NF-κB, which was revealed to promote the transition of tumor cells towards the mesenchymal phenotype through signaling from STAT3, TAZ, and C/EBPβ, potentially indicating the involvement of inflammation within EMT/PMT transitions [88].
A major hallmark of cancer is altered metabolism, namely a transition known as the Warburg effect, which describes a tumor shifting metabolism towards aerobic glycolysis in order to thrive despite hypoxic and acidic conditions [82,83]. Therefore, HIF-1α resides at the center of yet another malignant trend within GBM tumors. Here, HIF-1α is activated by translocating to the nucleus, leading to expression of genes associated with glycolysis such as glucose transporters 1 and 3 (GLUT1,3), while encouraging lactate buildup throughout the tumor [89]. Lactate propels the acidity of the GBM TME, which has immunosuppressive effects on the tumor by disabling CD8+ T-cells and NK cells, accomplishing another hallmark of cancer: avoiding immune destruction [89,90]. Additionally, lactate participates in epigenetic remodeling in GBM via lactylation of histones which can interfere with the process of phagocytosis, ultimately leading to increased proliferation of GBM cells [89]. It should be noted that GBM metabolism is also heterogeneous in itself, with proneural CSCs exhibiting features of normoxic metabolism, whilst mesenchymal CSCs rely more on glycolysis and altered metabolism. This trend is consistent with the signature aggression observed in mesenchymal phenotypes, as well as the expected clinical outcomes for both mesenchymal and proneural subtypes in GBM.
7. Conclusions
GBM is a highly heterogeneous, invasive, and aggressive form of brain cancer with intratumoral dynamics constantly evolving across tumor phenotypes and niches. This complex malignancy is characterized by numerous genetic, epigenetic, metabolic, cellular, and vascular abnormalities where hypoxia and highly regulated stem cell maintenance foster much of this diversity. Table 2 summarizes each property of GBM introduced in this review which contributes to its overall heterogeneity and plasticity.
While the works presented in this review will guide future research and illustrate great commitment to improving GBM patient care, little of this progress has translated clinically. Given the consistently poor survival outcomes of GBM patients worldwide, further research and therapeutic development is critical, and it has become clear that a change in thinking about how we do this research is needed. Fast, highly accurate, highly reproducible studies with beautiful error bars performed in uniform, well-behaved GBM cell models have not helped us to make meaningful advances in patient care. This is in part because many studies do not acknowledge or engage with the staggering cellular diversity and heterogeneity of this disease and the complex behaviors that emerge. Future work needs to be conceived and executed with a priority placed on highly accurate models and systems to maximize the potential success of future translation. Future directions of GBM research should further push the boundaries of the role of immune cells in therapeutic resistance, chromatin modifications contributing to CSC proliferation and overall tumor heterogeneity, metabolic abnormalities and specialized functionalities in each tumor niche, and active interactions with the normal cells of the patient. Gaining further understanding of the heterogeneity of GBM tumors and determining similarities between tumor phenotypes presents the opportunity to identify novel therapeutic targets and prognostic indicators to guide treatment and improve the overall survival of this deadly disease.

 ,
Lilly J. Speier
2,†
,
Lilly J. Speier
2,†