
Cancer Heterogeneity and Plasticity ISSN 2818-7792
Cancer Heterogeneity and Plasticity 2025;2(3):0013 | https://doi.org/10.47248/chp2502030013
Perspective Open Access
The Three Barriers Senescent Tumor Cells Must Overcome to Relapse
James G. Jackson


Academic Editor(s): Dean G. Tang
Received: May 28, 2025 | Accepted: Jul 27, 2025 | Published: Aug 4, 2025
© 2025 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Jackson JG. The Three Barriers Senescent Tumor Cells Must Overcome to Relapse. Cancer Heterog Plast. 2025;2(3):0013. https://doi.org/10.47248/chp2502030013
Tumor cells that enter senescence as a response to treatment can be permanently arrested or removed by the immune system, resulting in favorable patient outcomes. Alternatively, many studies have now shown that, in some tumors, the senescent program enables tumor cell survival, persistence, and eventually relapse, resulting in poor patient outcomes. Whether senescence is a positive or negative factor is dependent on a clonal population of cells overcoming three critical barriers. First, senescence must enable survival from the initial stress of treatment, such as DNA damage, by preventing apoptosis and/or mitotic catastrophe. Senescent cells are also frequently immunogenic, thus, a second barrier is the activation of programs of immune evasion, such as PD-L1 expression, that outweigh the immunogenic properties. Third, senescent cells must escape their rigid arrest to proliferate again. Studies over the years have experimentally addressed challenging questions related to relapse and senescence, but more research is needed, particularly in vivo. Here, we discuss critical studies investigating how tumor cells that enter senescence as a response to treatment overcome barriers to relapse.
KeywordsSenescence, relapse, cell cycle arrest, immunotherapy, immune evasion, p53, PD-L1
Cancer cells can respond to treatments in a variety of ways. Clearly, the most beneficial to the patient will be activation of extensive intrinsic cell death pathways coupled with surveillance by the immune system, resulting in complete tumor eradication and durable cure. This can and does occur in many tumors in response to therapies that induce cell death (such as chemotherapies) and/or activate the immune system (e.g., immune checkpoint inhibitors, or ICI). Of course, conventional and targeted therapies and the immune system frequently fail to eradicate the tumor, and surviving cells cause relapse and patient mortality.
Tumor cells rely on different strategies to survive and persist post treatment [1], including primary pharmacological resistance such as that mediated by drug efflux pumps [2]. Among strategies for cells that become stressed and damaged by treatments are various states of cell cycle arrest, including a diapause-like state [3,4] and, frequently, programs of senescence [5]. Both would result in a pathological stable disease or partial response [6]. Tumor volume decreases minimally and residual disease is frequently extensive. The senescence response to treatment occurs in breast and other cancers and has been investigated in animal models, and is the subject of this article.
Senescence response is best identified in cancers that are treated with neoadjuvant therapy, where the systemic treatment such as chemotherapy is given before the surgery, and thus the response to treatment can be characterized by molecular, transcriptomic, and immunohistochemical techniques in the surgical specimen. This has allowed senescence to be identified as a primary response in breast cancers that fail to undergo complete pathological response [7-10], and this has been followed up and studied in controlled experiments in animal models [10-12]. These studies have identified senescent cells using various means, including RNA expression data showing elevated cell cycle inhibitors, loss of Lamin B1 and cyclins/cdks, induction of p53 target genes as well as genes related to the senescence associated secretory phenotype, or SASP [10,12,13]. Studies have used staining for many of these same genes in histological sections of tumors, pre- and post-treatment, including Ki67, p21, p16, SASP genes, and Lamin B1 loss [7-9]. Lastly, studies have shown extensive positive staining for senescence marker SAβGal [14] when feasible [9,12]. In addition to breast, various reports have demonstrated therapy-induced senescence in patient samples or mouse models for prostate [15,16], pancreas [17], lung [18-20], mesothelioma [21], rectal [22], leukemia/lymphoma [23-25], head and neck squamous [26], and ovarian cancer [27]. As other tumor types are examined in the post-treatment period preceding relapse or eradication, it is likely that cells in a senescent-like state would be detected.
In between eradication and primary resistance is stable disease or partial response, where a tumor has responded to treatment, but viable cells remain. The tissue, cell, and stimulus specific regulation of senescent phenotypes will ultimately determine the fate of the tumor cell and the patient. One potential outcome is that any tumor cells that might remain after surgery are rigidly arrested, never proliferating again at a rate to produce a detectable relapse. It is also possible that these senescent cells may interact with the immune system to facilitate their removal over time, and it is known that senescent cells become immunogenic [10,16,28-30]. Lastly, entering a senescent state can introduce vulnerabilities to a second “senolytic” therapy. This strategy, particularly the targeting of the BCL2 family of antiapoptotic proteins, has had varying success [11,31-33], depending on the state of apoptotic priming of the senescent cells [34-36]. Thus, induction of senescence as a majority response in a tumor can effectuate a cure if the arrest is rigid, the immune system is activated, and/or senolytic drugs can be used to remove them.
The extent of senescence within the tumor is a critical factor in the response. Senescence can vary in depth and expression of other various phenotypes [37]. If a tumor loses only a small amount of volume post-treatment but then remains stable, the extent of the senescent arrest is likely widespread. If the tumor growth stalls briefly and then resumes, it is fair to speculate that senescence was either not induced in many cells that also resisted cell death, or that the arrest was shallow and easily exited [37]. Too frequently, this is the case, and the senescence response by a tumor actually prevents realizing a cure, ultimately promoting relapse and patient mortality. This is glaringly evident in breast cancer, where p53 wild-type tumors are the most difficult to eradicate with chemotherapy [38-40] and survival of these patients is much shorter than those with p53 mutant tumors that are far less likely to enter senescence after treatment [41-44]. Thus, the induction of senescence is heterogeneous across the population of tumor cells, with some cells sustaining more or less damage, during different phases of the cell cycle, and in different immune and tumor microenvironment contexts. This heterogeneity allows selection of the fittest clones over the next phases toward relapse.
Here, we discuss three layers of selection that a tumor cell that has entered senescence must overcome to cause eventual relapse.
The first requirement for the survival of a tumor cell post-treatment is avoiding cell death in response to toxic stresses, including DNA damage, mitotic spindle poisons, and therapies that target oncogenic drivers (Figure 1A-B). The state of senescence can facilitate and enable this survival by multiple mechanisms. Mouse models of breast cancer have shown that TP53 mutant tumor cells that fail to arrest following DNA damaging chemotherapy continue through S-phase and mitosis, ultimately undergoing mitotic catastrophe, and perhaps other modes of cell death [12]. TP53 wild type breast tumors, however, enter senescence and are not eradicated by the chemotherapy [12,38-40]. In fact, in many tumors, a majority of cells will not be eliminated by the initial chemotherapy, and instead, senescence is the primary outcome [45-47]. This is not necessarily a case of rare persisters. In breast cancers, p53 transactivates many genes related to apoptosis, but cell cycle arrest and senescence is the outcome for many cells. Cell types that are inherently resistant to apoptosis are not “primed” to undergo apoptosis following p53-mediated expression of pro-apoptotic genes such as Puma, Noxa, and Bax [48,49]. Residual disease is often quite extensive in patients or models systems with TP53 wild-type tumors [12,38-40].

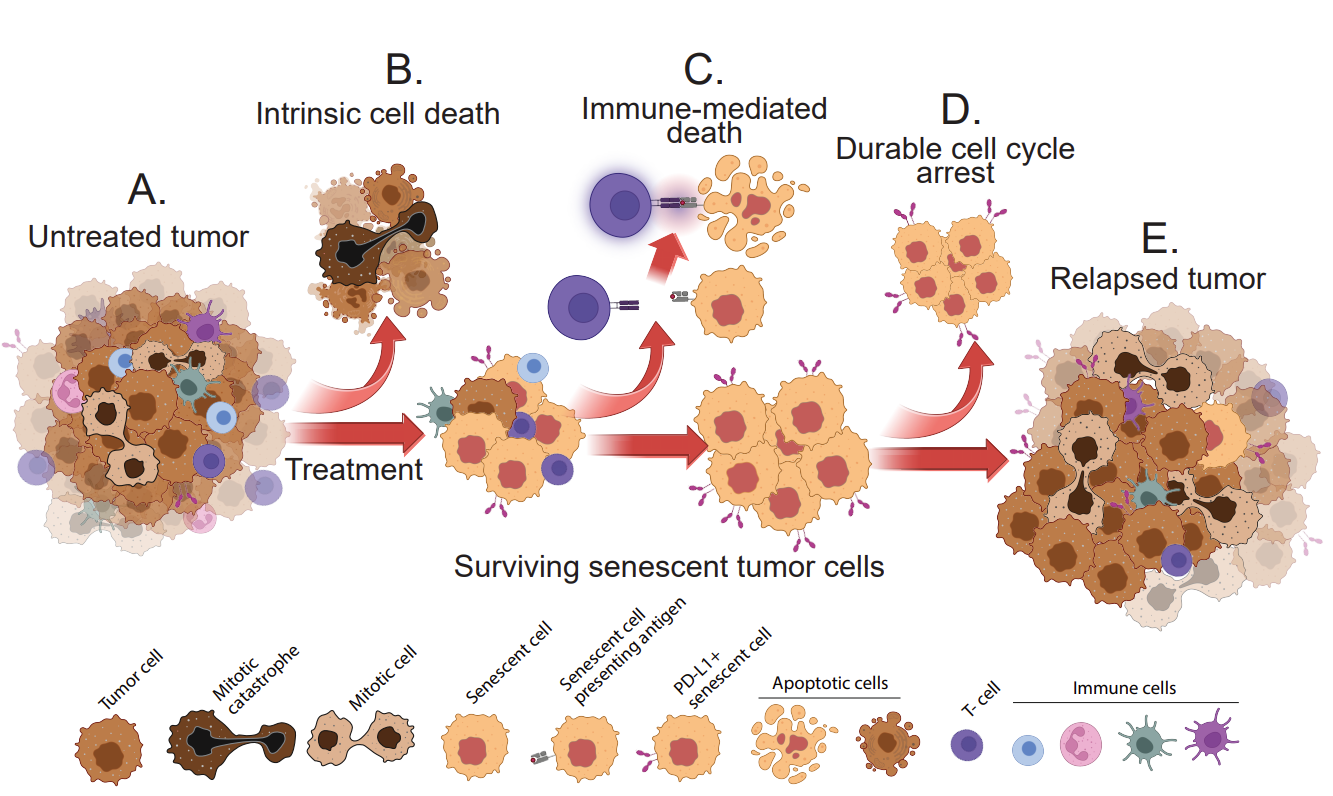
Figure 1. Three layers of selection must be overcome for senescent tumor cells to cause relapse. (A) An untreated, growing tumor is comprised of dividing tumor cells and various stromal/immune cell types. (B) Following treatment, such as with a chemotherapy drug, apoptosis or mitotic catastrophe will result in intrinsic cell death, but cells that enter senescence frequently survive. C) The cells that survive apoptosis must then evade the immune system, including cytotoxic T-cells. (D) Senescent tumor cells that remain viable and persist, can be permanently arrested. (E) Some tumor cells that expressed senescent phenotypes can re-enter the cell cycle, begin proliferating again, and cause relapse.
As mentioned above, reliance on BCL2 family members such as BCL-XL to survive, however, does create a vulnerability to “senolytic” agents that block BCL2 family members [11,31,34]. Thus, these agents might be used to improve response to treatment by throwing cells into apoptosis that are surviving in senescence [50]. Pre-clinical data suggest that indeed, some fraction of the cells in the tumor can be eliminated, improving response and survival in mouse models, but there remain enough cells in the heterogeneous population that resist and survive [11, 51]. Further, cell lines exhibit an array of sensitivities to the various BCL2 family targeting drugs [11,51]. One mechanism employed by cells that resist BCL-XL inhibition is activity of another BCL2 family member, MCL-1, shown initially in breast [11] and then prostate [32] and melanoma [52]. MCL1 can be targeted by a second drug, and more senescent cells in the population can be killed, but this combination is probably too toxic to provide clinical benefit.
Breast and other tumor cell types that have survived treatment by entering senescence express an array of genes related to macrophages and dendritic cells, and acquire the ability to engulf and break down various large targets, including apoptotic cells, healthy proliferating cells, and other senescent cancer cells [53,54]. Further, senescent tumor cells have increased capacity for autophagy [55]. It is reasonable to hypothesize that the ability to acquire nutrients via engulfment or autophagy would enhance survival of senescent cells, but more research is needed to understand this definitively, especially in vivo where very little is known.
Among the large population of tumor cells that become senescent after treatment are those that express differing levels and combinations of immune modulatory genes [56]. These include cytokines and chemokines of the SASP [57], as well as checkpoint ligands and other immune co-stimulatory or inhibitory genes [46,58]. This creates a next layer of selection for tumor cells that will reach relapse: evade the immune system (Figure 1C).
Recent studies make clear that genes that promote immune surveillance are expressed by senescent cells. These include antigen presentation genes and various cytokines and chemokines that can attract and activate T-cells [10,28,29,59] and NK cells [20,60-62]. For a tumor cell to survive, these immunity promoting phenotypes must be counteracted by immune inhibitory pathways, and, indeed, senescent tumor cells simultaneously express an array of genes that suppress the immune system. These include clinically important genes such as PD-L1, PD-L2, and Galectin 9 [10,63,64]. In some tumor types such as breast, it is clear the ultimate outcome for tumors that enter senescence is relapse. In these cancers, the balance of immune regulation must be tilted toward evasion. Expression of PD-L1/PD-L2 and other suppressive pathways “wins” over antigen presentation and immunogenicity, thus the tumor survives to relapse.
Mechanisms of immune evasion, however, can be targeted by immunotherapies such as immune checkpoint inhibitors (ICI), including anti PD-L1 and PD-L2 antibodies [10,64], as well as strategies to increase immunogenicity of tumor cells [65]. The blocking of the negative signal transmitted from PD-L1 and/or PD-L2 on tumor cells to the PD1 expressed on T cells, combined with the expression of antigen presentation and costimulatory genes, can result in a robust response to ICI by senescent tumor cells [10,64]. There is also, however, redundancy in the system. Senescent cells can express many immune modulatory genes, including various checkpoint ligands and cytokines [10]. Thus, blocking one may not be enough to overcome the sum of negative signals being generated by the senescent cell.
The final layer of selection in the path to relapse is the ability of a cell to emerge from senescent arrest to proliferate again and create a growing tumor (Figure 1D-E). Relatively little is known about these cells for reasons of intractability in their study and characterization. Cells with an evident senescent morphology have been observed to restart proliferation in vitro [51,66-69]. In vivo studies have correlated the presence of senescent cells in a tumor at one time point with proliferating cells at a later time point, but the question of whether the proliferating cell had at one time harbored some number of senescence properties is not clear. This conundrum was recently addressed in a model of tumor suppression, not therapy induced senescence. The authors generated hepatocytes with a Cdkn1a promoter driving Cre recombinase that could convert a tdTomato expressing allele to a GFP allele. This, in effect, would permanently mark with GFP any cell that had expressed the p53 target and senescence gene Cdkn1a. They found that hepatocytes that expressed Cdkn1a in response to metabolic stress indeed proliferated again as transformed carcinoma cells [70]. This is a powerful model for testing emergence from senescence, however, the Cdkn1a promoter is activated by p53 within hours after stress, long before cells become senescent. It is possible these Cdkn1a positive cells did not co-express various other markers and effectors of senescence.
In another recent report, in vitro experiments showed that in a population of cells that were treated, the surviving cells were mostly positive for senescence markers and morphology, and these cells did begin dividing again. The “repopulating” cells were sensitive to further treatment, and were very similar to untreated cells in gene expression and single cell population composition [51]. It is not entirely clear from the study design the precise origin or nature of these repopulating cells. This would include characterization of the specific cells from which the repopulating cells were derived or if their program of arrest had any defining characteristics such as chromatin structure [71,72] or cytoplasmic chromatin activating c-GAS/Sting [73], which both can regulate the depth and quality of senescence in normal cells such as fibroblasts. In another recent study, the authors showed that chemotherapy treated, immortalized mammary epithelial cell line MCF-10A expressed many typical markers of senescence with graded intensity rather than a binary state. The more intensely positive for features such as SAβGal, the longer the duration before cell cycle re-entry [74].
In vivo evidence that a “previously therapy-induced senescent cell” (how this is defined varies, and is important) can proliferate again to repopulate the relapsed tumor, has not been rigorously demonstrated. It is possible that in some tumor types senescence is more rigid, more locked into a chromatin state. As discussed recently, it is likely that in a heterogeneous tumor cell population, various cells display a continuum across the arrest spectrum that includes cells that readily escape and proliferate again and those that are permanently arrested [37,74].
Whether the senescence response to treatment is favorable or detrimental to the patient’s outcome is dependent on the ability of senescent cells to overcome three barriers to relapse: apoptosis, immune surveillance, and durable cell cycle arrest (Figure 1). Tumor cells in a heterogeneous population will vary widely in the degree to which each senescent phenotype is expressed. Ultimately, as selective pressures are placed on this population by the treatment, the immune system, and the depth of the arrest, it will take only a small number of cells to survive and emerge to cause relapse. Targeting these cells at any of these 3 selection bottlenecks, such as by using senolytics or immunotherapies, will likely provide benefit.
Not applicable.
Not applicable.
Not applicable.
National Institutes of Health (R01CA259001).
James G. Jackson is a member of the Editorial Board of the journal Cancer Heterogeneity and Plasticity. The author was not involved in the journal’s review of or decisions related to this manuscript. The author has declared that no other competing interests exist.
The author acknowledges BioRender was used to create the figure.
| 1. | Russo M, Chen M, Mariella E, Peng H, Rehman SK, Sancho E, et al. Cancer drug-tolerant persister cells: from biological questions to clinical opportunities. Nat Rev Cancer. 2024;24(10):694-717. [Google Scholar] [CrossRef] |
| 2. | Sharom FJ. ABC Multidrug Transporters: Structure, Function and Role in Chemoresistance. Pharmacogenomics. 2008;9(1):105-127. [Google Scholar] [CrossRef] |
| 3. | Rehman SK, Haynes J, Collignon E, Brown KR, Wang Y, Nixon AML, et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell. 2021;184(1):226-242.e221. [Google Scholar] [CrossRef] |
| 4. | Dhimolea E, de Matos Simoes R, Kansara D, Al'Khafaji A, Bouyssou J, Weng X, et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell. 2021;39(2):240-256.e211. [Google Scholar] [CrossRef] |
| 5. | Schmitt CA. Persistence and/or Senescence: Not So Lasting at Last? Cancer Res. 2025;85(1):7-9. [Google Scholar] [CrossRef] |
| 6. | Song K-X, Wang J-X, Huang D. Therapy-induced senescent tumor cells in cancer relapse. J Natl Cancer Cent. 2023;3(4):273-278. [Google Scholar] [CrossRef] |
| 7. | Saleh T, Alhesa A, Al-Balas M, Abuelaish O, Mansour A, Awad H, et al. Expression of therapy-induced senescence markers in breast cancer samples upon incomplete response to neoadjuvant chemotherapy. Biosci Rep. 2021;41(5). [Google Scholar] [CrossRef] |
| 8. | El-Sadoni M, Shboul SA, Alhesa A, Shahin NA, Alsharaiah E, Ismail MA, et al. A three-marker signature identifies senescence in human breast cancer exposed to neoadjuvant chemotherapy. Cancer Chemother Pharmacol. 2023;91(4):345-360. [Google Scholar] [CrossRef] |
| 9. | te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62(6):1876-1883. [Google Scholar] |
| 10. | Shahbandi A, Chiu F-Y, Ungerleider NA, Kvadas R, Mheidly Z, Sun MJS, et al. Breast cancer cells survive chemotherapy by activating targetable immune-modulatory programs characterized by PD-L1 or CD80. Nat Cancer. 2022;3(12):1513-1533. [Google Scholar] [CrossRef] |
| 11. | Shahbandi A, Rao SG, Anderson AY, Frey WD, Olayiwola JO, Ungerleider NA, et al. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020;27(11):3097-3116. [Google Scholar] [CrossRef] |
| 12. | Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, et al. p53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell. 2012;21(6):793-806. [Google Scholar] [CrossRef] |
| 13. | Tonnessen-Murray C, Ungerleider NA, Rao SG, Wasylishen AR, Frey WD, Jackson JG. p53 Mediates Vast Gene Expression Changes That Contribute to Poor Chemotherapeutic Response in a Mouse Model of Breast Cancer. Transl Oncol. 2018;11(4):930-940. [Google Scholar] [CrossRef] |
| 14. | Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363-9367. [Google Scholar] [CrossRef] |
| 15. | Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853-2868. [Google Scholar] [CrossRef] |
| 16. | Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9(1):75-89. [Google Scholar] [CrossRef] |
| 17. | Ruscetti M, Morris JPt, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2020;181(2):424-441.e421. [Google Scholar] [CrossRef] |
| 18. | Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65(7):2795-2803. [Google Scholar] [CrossRef] |
| 19. | Domen A, Deben C, De Pauw I, Hermans C, Lambrechts H, Verswyvel J, et al. Prognostic implications of cellular senescence in resected non-small cell lung cancer. Transl Lung Cancer Res. 2022;11(8):1526-1539. [Google Scholar] [CrossRef] |
| 20. | Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362(6421):1416-1422. [Google Scholar] [CrossRef] |
| 21. | Sidi R, Pasello G, Opitz I, Soltermann A, Tutic M, Rehrauer H, et al. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: an exploratory analysis. Eur J Cancer. 2011;47(2):326-332. [Google Scholar] [CrossRef] |
| 22. | Tato-Costa J, Casimiro S, Pacheco T, Pires R, Fernandes A, Alho I, et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin Colorectal Cancer. 2016;15(2):170-178.e173. [Google Scholar] [CrossRef] |
| 23. | Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021;11(6):1542-1561. [Google Scholar] [CrossRef] |
| 24. | Belenki D, Richter-Pechanska P, Shao Z, Bhattacharya A, Lau A, Nabuco Leva Ferreira de Freitas JA, et al. Senescence-associated lineage-aberrant plasticity evokes T-cell-mediated tumor control. Nat Commun. 2025;16(1):3079. [Google Scholar] [CrossRef] |
| 25. | Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335-346. [Google Scholar] [CrossRef] |
| 26. | Schoetz U, Klein D, Hess J, Shnayien S, Spoerl S, Orth M, et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. 2021;12(12):1162. [Google Scholar] [CrossRef] |
| 27. | Paffenholz SV, Salvagno C, Ho Y-J, Limjoco M, Baslan T, Tian S, et al. Senescence induction dictates response to chemo- and immunotherapy in preclinical models of ovarian cancer. Proc Natl Acad Sci U S A. 2022;119(5):e2117754119. [Google Scholar] [CrossRef] |
| 28. | Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, et al. Cellular senescence is immunogenic and promotes anti-tumor immunity. Cancer Discov. 2022;3(2):410-431. [Google Scholar] [CrossRef] |
| 29. | Chen HA, Ho YJ, Mezzadra R, Adrover JM, Smolkin R, Zhu C, et al. Senescence rewires microenvironment sensing to facilitate anti-tumor immunity. Cancer Discov. 2022. [Google Scholar] [CrossRef] |
| 30. | Munoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. 2019;5. [Google Scholar] [CrossRef] |
| 31. | Saleh T, Carpenter VJ, Tyutyunyk-Massey L, Murray G, Leverson JD, Souers AJ, et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-XL -BAX interaction. Mol Oncol. 2020;14(10):2504-2519. [Google Scholar] [CrossRef] |
| 32. | Troiani M, Colucci M, D’Ambrosio M, Guccini I, Pasquini E, Varesi A, et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat Commun. 2022;13(1):2177. [Google Scholar] [CrossRef] |
| 33. | Wang L, Jin H, Jochems F, Wang S, Lieftink C, Martinez IM, et al. cFLIP suppression and DR5 activation sensitize senescent cancer cells to senolysis. Nat Cancer. 2022;3(11):1284-1299. [Google Scholar] [CrossRef] |
| 34. | MacDonald JA, Bradshaw GA, Jochems F, Bernards R, Letai A. Apoptotic priming in senescence predicts specific senolysis by quantitative analysis of mitochondrial dependencies. Cell Death Differ. 2025;32(5):802-817. [Google Scholar] [CrossRef] |
| 35. | López J, Llop-Hernández À, Verdura S, Serrano-Hervás E, Martinez-Balibrea E, Bosch-Barrera J, et al. Mitochondrial priming and response to BH3 mimetics in “one-two punch” senogenic-senolytic strategies. Cell Death Discov. 2025;11(1):91. [Google Scholar] [CrossRef] |
| 36. | Jochems F, Baltira C, MacDonald JA, Daniels V, Mathur A, de Gooijer MC, et al. Senolysis by ABT-263 is associated with inherent apoptotic dependence of cancer cells derived from the non-senescent state. Cell Death Differ. 2025;32(5):855-865. [Google Scholar] [CrossRef] |
| 37. | Reimann M, Lee S, Schmitt CA. Cellular senescence: Neither irreversible nor reversible. J Exp Med. 2024;221(4):e20232136. [Google Scholar] [CrossRef] |
| 38. | Bertheau P, Plassa F, Espie M, Turpin E, de Roquancourt A, Marty M, et al. Effect of mutated TP53 on response of advanced breast cancers to high-dose chemotherapy. Lancet. 2002;360(9336):852-854. [Google Scholar] [CrossRef] |
| 39. | Bertheau P, Turpin E, Rickman DS, Espie M, de Reynies A, Feugeas JP, et al. Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin-cyclophosphamide regimen. PLoS Med. 2007;4(3):e90. [Google Scholar] [CrossRef] |
| 40. | Wang Y, Xu Y, Chen J, Ouyang T, Li J, Wang T, et al. TP53 mutations are associated with higher rates of pathologic complete response to anthracycline/cyclophosphamide-based neoadjuvant chemotherapy in operable primary breast cancer. Int J Cancer. 2016;138(2):489-496. [Google Scholar] [CrossRef] |
| 41. | Ungerleider NA, Rao SG, Shahbandi A, Yee D, Niu T, Frey WD, et al. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res. 2018;20(1):115. [Google Scholar] [CrossRef] |
| 42. | Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer. 2020;6(2):98-110. [Google Scholar] [CrossRef] |
| 43. | Kim JY, Lee E, Park K, Park WY, Jung HH, Ahn JS, et al. Clinical implications of genomic profiles in metastatic breast cancer with a focus on TP53 and PIK3CA, the most frequently mutated genes. Oncotarget. 2017;8(17):27997-28007. [Google Scholar] [CrossRef] |
| 44. | Nakamura Y, Oshima K, Naoi Y, Nakayama T, Kim SJ, Shimazu K, et al. 14-3-3sigma expression is associated with poor pathological complete response to neoadjuvant chemotherapy in human breast cancers. Breast Cancer Res. 2012;134(1):229-236. [Google Scholar] [CrossRef] |
| 45. | Saleh T, Greenberg EF, Faber AC, Harada H, Gewirtz DA. A Critical Appraisal of the Utility of Targeting Therapy-Induced Senescence for Cancer Treatment. Cancer Res. 2025;85(10):1755-1768. [Google Scholar] [CrossRef] |
| 46. | Zhou L, Ma B, Ruscetti M. Cellular senescence offers distinct immunological vulnerabilities in cancer. Trends Cancer. 2025;11(4):334-350. [Google Scholar] [CrossRef] |
| 47. | Fitsiou E, Soto-Gamez A, Demaria M. Biological functions of therapy-induced senescence in cancer. Semin Cancer Biol. 2022;81:5-13. [Google Scholar] [CrossRef] |
| 48. | Potter DS, Letai A. To Prime, or Not to Prime: That Is the Question. Cold Spring Harb Symp Quant Biol. 2016;81:131-140. [Google Scholar] [CrossRef] |
| 49. | Sánchez-Rivera FJ, Ryan J, Soto-Feliciano YM, Clare Beytagh M, Xuan L, Feldser DM, et al. Mitochondrial apoptotic priming is a key determinant of cell fate upon p53 restoration. Proc Natl Acad Sci U S A. 2021;118(23):e2019740118. [Google Scholar] [CrossRef] |
| 50. | McHugh D, Durán I, Gil J. Senescence as a therapeutic target in cancer and age-related diseases. Nat Rev Drug Discov. 2025;24(1):57-71. [Google Scholar] [CrossRef] |
| 51. | Bajtai E, Kiss C, Bakos É, Langó T, Lovrics A, Schád É, et al. Therapy-induced senescence is a transient drug resistance mechanism in breast cancer. Mol Cancer. 2025;24(1):128. [Google Scholar] [CrossRef] |
| 52. | Kohli J, Ge C, Fitsiou E, Doepner M, Brandenburg SM, Faller WJ, et al. Targeting anti-apoptotic pathways eliminates senescent melanocytes and leads to nevi regression. Nat Commun. 2022;13(1):7923. [Google Scholar] [CrossRef] |
| 53. | Frey WD, Anderson AY, Lee H, Nguyen JB, Cowles EL, Lu H, et al. Phosphoinositide species and filamentous actin formation mediate engulfment by senescent tumor cells. PLoS Biol. 2022;20(10):e3001858. [Google Scholar] [CrossRef] |
| 54. | Tonnessen-Murray CA, Frey WD, Rao SG, Shahbandi A, Ungerleider NA, Olayiwola JO, et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J Cell Biol. 2019;218(11):3827-3844. [Google Scholar] [CrossRef] |
| 55. | Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat Res. 2020;194(2):103-115. [Google Scholar] [CrossRef] |
| 56. | Zingoni A, Antonangeli F, Sozzani S, Santoni A, Cippitelli M, Soriani A. The senescence journey in cancer immunoediting. Mol Cancer. 2024;23(1):68. [Google Scholar] [CrossRef] |
| 57. | Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125-2136. [Google Scholar] [CrossRef] |
| 58. | Chibaya L, Snyder J, Ruscetti M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin Cancer Biol. 2022;86(3):827-845. [Google Scholar] [CrossRef] |
| 59. | Liu Y, Pagacz J, Wolfgeher DJ, Bromerg KD, Gorman JV, Kron SJ. Senescent cancer cell vaccines induce cytotoxic T cell responses targeting primary tumors and disseminated tumor cells. J Immunotherap Cancer. 2023;11(2):e005862. [Google Scholar] [CrossRef] |
| 60. | Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210(10):2057-2069. [Google Scholar] [CrossRef] |
| 61. | Antonangeli F, Soriani A, Ricci B, Ponzetta A, Benigni G, Morrone S, et al. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. OncoImmunology. 2016;5(10):e1218105. [Google Scholar] [CrossRef] |
| 62. | Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503-3511. [Google Scholar] [CrossRef] |
| 63. | Hwang HJ, Kang D, Shin J, Jung J, Ko S, Jung KH, et al. Therapy-induced senescent cancer cells contribute to cancer progression by promoting ribophorin 1-dependent PD-L1 upregulation. Nat Commun. 2025;16(1):353. [Google Scholar] [CrossRef] |
| 64. | Chaib S, Lopez-Dominguez JA, Lalinde-Gutierrez M, Prats N, Marin I, Boix O, et al. The efficacy of chemotherapy is limited by intratumoral senescent cells expressing PD-L2. Nat Cancer. 2024;5(3):448-462. [Google Scholar] [CrossRef] |
| 65. | Chibaya L, DeMarco KD, Lusi CF, Kane GI, Brassil ML, Parikh CN, et al. Nanoparticle delivery of innate immune agonists combined with senescence-inducing agents promotes T cell control of pancreatic cancer. Science Translational Medicine. 2024;16(762):eadj9366. [Google Scholar] [CrossRef] |
| 66. | Saleh T. Therapy-induced senescence is finally escapable, what is next? Cell Cycle. 2024;23(6):713-721. [Google Scholar] [CrossRef] |
| 67. | Miller D, Kerkhofs K, Abbas-Aghababazadeh F, Madahar SS, Minden MD, Hébert J, et al. Heterogeneity in leukemia cells that escape drug-induced senescence-like state. Cell Death Dis. 2023;14(8):503. [Google Scholar] [CrossRef] |
| 68. | Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol. 2019;162:202-212. [Google Scholar] [CrossRef] |
| 69. | Fleury H, Malaquin N, Tu V, Gilbert S, Martinez A, Olivier M-A, et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun. 2019;10(1):2556. [Google Scholar] [CrossRef] |
| 70. | Gu L, Zhu Y, Nandi SP, Lee M, Watari K, Bareng B, et al. FBP1 controls liver cancer evolution from senescent MASH hepatocytes. Nature. 2025;637(8045):461-469. [Google Scholar] [CrossRef] |
| 71. | Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22(16):4212-4222. [Google Scholar] [CrossRef] |
| 72. | Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703-716. [Google Scholar] [CrossRef] |
| 73. | Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402-406. [Google Scholar] [CrossRef] |
| 74. | Ashraf HM, Fernandez B, Spencer SL. The intensities of canonical senescence biomarkers integrate the duration of cell-cycle withdrawal. Nat Commun. 2023;14(1):4527. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy





Article Comments