Abstract
The incidence and mortality of prostate cancer (PCa) are increasing worldwide, affecting the health of millions of men. Metastatic PCa (mPCa) represents a critical challenge in terms of clinical treatment because of its aggressive invasiveness and metastatic potential, making it a main cause of death in this population. Although bone metastasis is more common in PCa patients, nonbone metastases, such as liver, lung, and brain metastases, are typically associated with a poorer prognosis. However, the mechanisms underlying nonbone metastasis in PCa are still not fully understood. This review summarizes the characteristics of tumor cells, the tumor microenvironment, research models, and diagnostic and therapeutic approaches for nonbone metastasis in PCa, with a particular focus on liver metastasis, and proposes feasible directions for future research.
Keywords
Prostate cancer, cancer metastasis, nonbone metastasis, tumor microenvironment, neuroendocrine
1. Introduction
Prostate cancer (PCa) is a malignant tumor to which men are highly susceptible. The incidence and mortality of PCa are increasing worldwide [1–3]. It ranks first in new cancer cases among men and second in cancer-related deaths, highlighting its heavy disease burden [1,2]. The progression of PCa is characterized by multiple stages [4]. The growth of PCa relies on androgen, making androgen deprivation therapy (ADT) a critical treatment for PCa [5]. For patients with localized, nonmetastatic disease, primary treatments include radical prostatectomy or radiotherapy, with the selective use of transient ADT. However, most patients show increased disease burden after primary treatment, characterized by rising PSA levels and/or radiographic progression. Therefore, continued ADT treatment is warranted. When ADT fails, the resulting castration-resistant PCa (CRPC) is treated with next-generation androgen receptor pathway inhibitors (ARPIs). Nevertheless, the majority of patients experience disease recurrence after ARPIs treatment, ultimately leading to death in these CRPC patients [4,6]. CRPC not only develops resistance to ADT but also exhibits further invasive proliferation, leading to metastatic CRPC (mCRPC), which is the primary cause of death in this population [4]. In China, approximately 28% of newly diagnosed patients have already developed distant metastasis, and this high metastatic rate leads to a poor prognosis and a significantly reduced survival rate [7]. The 5-year survival rate for patients with localized PCa without metastasis is 90–99%, whereas that for mCRPC patients is only 30–40%, underscoring the malignancy of mCRPC [2].
Regarding the organotropism of cancer metastasis, the classic "seed and soil" theory was proposed as early as 1889 [8,9]. PCa also exhibits significant organotropism, with bones and lymph nodes being the most common metastatic sites, followed by nonbone organs such as the liver, lungs, and brain [4,10]. Among various metastatic sites, nonbone metastases are associated with shorter survival periods and poorer prognoses than are bone metastasis [11]. The molecular mechanisms of PCa bone metastasis have been extensively studied [12,13]. However, the mechanisms of nonbone metastasis remain unclear, and owing to its insidious nature, early diagnosis is often challenging, leading to a poor prognosis. Moreover, there is still a lack of specific therapeutic strategies for PCa nonbone metastasis.
In recent years, molecular subtype studies based on androgen receptor (AR) and neuroendocrine (NE) gene expression features have provided new insights into the heterogeneity of mCRPC. According to AR and NE marker expression patterns, mCRPC can be classified into five clusters: AR+NE- tumors (ARPC), ARlowNE- tumors (ARLPC), AR+NE+ tumors (AMPC), AR-NE+ tumors (NEPC), and AR-NE- tumors (DNPC) [14]. Among these subtypes, NEPC and DNPC exhibit increased migration, invasion, and metastatic capabilities, with significant differences in molecular characteristics, metastatic tendencies, and treatment responses. The proportions of NEPC and DNPC increase after ADT, and these tumors develop resistance to ARPIs (e.g., enzalutamide and abiraterone acetate [15–20]). Therefore, further research into the molecular mechanisms and biological behaviors of these PCa subtypes will not only help elucidate the heterogeneity of mCRPC but also provide a critical foundation for developing precision treatment strategies for nonbone metastatic PCa (mPCa).
This review summarizes the mechanisms of nonbone metastasis in PCa, focusing on the tumor characteristics and microenvironmental features of nonbone mPCa, particularly in the case of liver metastasis. Current research models and clinical diagnostic and therapeutic strategies are also reviewed with the aim of providing new insights and theoretical foundations for basic research and clinical treatment.
2. Hallmarks of Nonbone mPCa
Most PCa patients progress to mCRPC after ADT treatment [6]. At this stage, PCa cells not only exhibit aggressive proliferation capabilities but are also highly prone to distant metastasis. Compared with localized PCa, mCRPC shows significant differences in genomic characteristics [21–24]. Germline mutations in DNA damage repair (DDR) genes and mismatch repair-related genes increase the likelihood of developing PCa [25,26]. Localized PCa typically has a low mutational burden [23,24]. However, mCRPC patients exhibit significantly increased mutational burdens and copy number alteration frequencies, reflecting therapy-induced genomic remodeling that results in the accumulation of numerous mutations [22,27]. For example, BRCA2 mutations are found in 12.5% of mCRPC patients, whereas these mutations are rare in patients with localized PCa [22,27,28]. Certain nonbone metastatic sites present distinct characteristics. Compared with PCa with metastasis to other sites, brain metastatic PCa shows relative enrichment of homologous recombination deficiency (HRD) mutations [29]. In one study, patients with brain metastases from PCa were found to have at least one mutation or somatic copy number alterations in homologous recombination repair genes. Compared with high-grade primary prostate tumors in a TCGA cohort, primary tumors from patients with brain metastases presented higher levels of HRD-related mutations [29]. Therefore, mutations in DDR and mismatch repair-related genes are closely associated with brain metastatic PCa and could serve as risk stratification biomarkers for PCa progression to brain metastasis. Brain metastatic PCa exhibits a CpG island hypermethylation phenotype, but hypomethylation is observed at promoters of neuroactive ligand‒receptor interactions and cell adhesion molecules (e.g., GABRB3, CLDN8, and CLDN4) [30]. This methylation pattern suggests that tumor cells undergo epigenetic reprogramming to adapt to the brain microenvironment. Additionally, brain metastatic PCa shows greater structural alterations in the genome, with chromothripsis and chromoplexy events predominating [31].
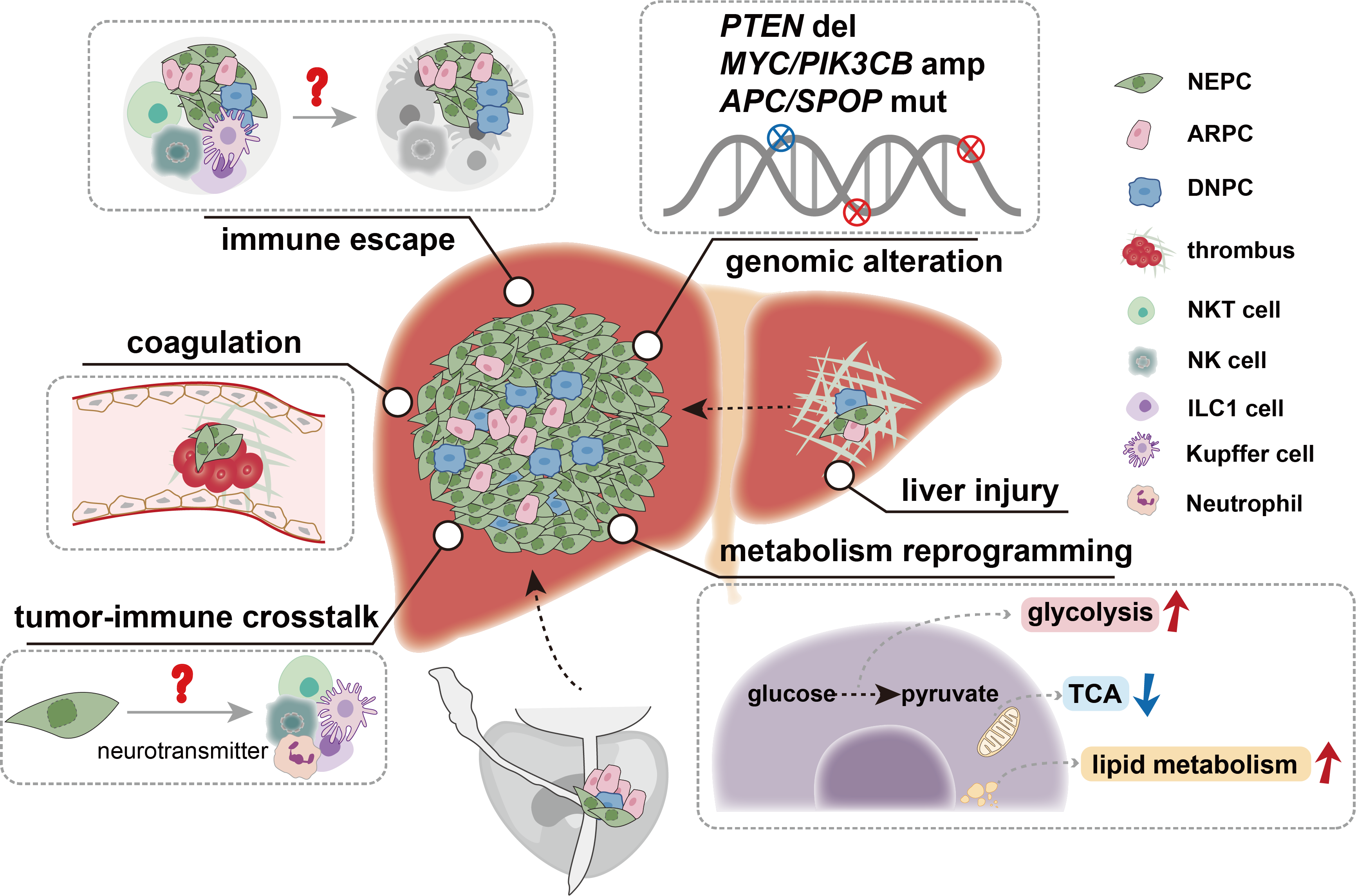
PTEN, the most frequently mutated tumor suppressor gene, plays a critical role in PCa development [4]. The loss of PTEN is significantly greater in mCRPC (>40%) than in localized and metastatic castration-sensitive PCa (mCSPC) tumors (12–17%) [21,22]. Genomic analysis of PCa tissue-specific metastasis revealed that mutations related to liver metastasis were more enriched in PTEN deletion, MYC amplification, and PIK3CB amplification than those related to metastasis to other sites [32]. However, no point mutations were found to be significantly associated with liver metastasis compared with metastasis to other sites [32]. Another study revealed that PTEN loss, as well as mutations in APC and SPOP, was more frequently enriched in the NEPC subtype, with a propensity for liver metastasis [33]. NEPC exhibits high migratory and invasive capabilities and plays a significant role in PCa liver metastasis [15,20]. Compared with other PCa subtypes, NEPC frequently harbors mutations in RB1 and TP53 [16,34,35]. Mutations in Rb1 or Trp53 alone are insufficient to form PCa, but their comutation leads to PCa with a neuroendocrine phenotype, which is prone to metastasis to the liver, lungs and other sites [36,37]. Studies have shown that in Pten-mutant mouse models of PCa, Rb1 loss promotes lineage plasticity and metastasis, whereas additional Trp53 loss induces resistance to ADT, explaining the ADT resistance and high visceral metastatic potential of NEPC [35]. A study of a patient with dural and brain metastases revealed that the dural metastasis coexpressed AR and NE markers, presenting as AMPC, whereas the brain metastasis exhibited NEPC characteristics. The AMPC-like metastasis showed elevated FOXA1 activity, whereas the brain NEPC-like metastasis exhibited increased expression of HOXC10, NFYB, and OTX2, indicating that neuroendocrine features may also contribute to the propensity for brain metastasis [38]. According to Stand Up To Cancer (SU2C) data, the proportion of AR-negative and NEPC patients with lung metastasis (14.3%) is lower than that of patients with liver metastasis (28.1%) but higher than that of patients with metastasis to other sites, such as the lymph nodes (6%) and bones (0.6%) [34,39].
DNPC is a unique mCRPC subtype accounting for up to 10%–25% of metastatic CRPC cases, especially after treatment with second-generation ARPIs [18]. The transcription factor KLF5 is upregulated in DNPC through demethylation, significantly enhancing tumor invasiveness [19]. While DNPC still demonstrates a notable incidence of liver metastasis and bone metastasis in most datasets, a subset of patient exhibits lung metastasis [14,16,40]. In DNPC, abnormal activation of the HGF/MET pathway significantly enhances Wnt/β-catenin signaling, further promoting DNPC formation and lung metastasis [41]. Additionally, the elevated activity of XPO1 and ribosomal proteins in DNPC suggests that inhibiting the XPO1 and ribosomal pathways may represent new therapeutic strategies for DNPC [41]. Loss of Kmt2c promotes the formation of PCa lung metastases. On the one hand, Kmt2c deficiency reduces the expression of the cell cycle inhibitor p16INK4A; on the other hand, it enhances Myc signaling and upregulates genes in the 5qE1 region (e.g., Odam and Cabs1), promoting lung metastasis [42,43]. Recent studies have highlighted the role of KMT2C in DNPC regulation. ADT induces KMT2C binding to enhancers of AR-regulated genes, preserving the adenocarcinoma lineage. KMT2C inactivation reduces ASPP2 expression, triggering Np63-dependent transdifferentiation. This DNPC transformation maintains fatty acid synthesis through Np63-mediated SREBP1c transactivation and promotes DNPC growth via HRAS palmitoylation and MAPK signaling activation [44]. These findings underscore KMT2C as an epigenetic checkpoint in DNPC development and provide new insights into the epigenetic mechanisms underlying PCa, particularly lung metastatic DNPC. In a subset of DNPC patients, tumor cells exhibit a squamous cell carcinoma phenotype [45]. The development and metastatic mechanisms of this phenotype, as well as its similarities to pulmonary squamous cell carcinoma, warrant further investigation.
Metabolic reprogramming, a hallmark of cancer, plays a pivotal role in tumor metastasis by enabling cancer cells to adapt to diverse microenvironments and overcome metabolic stress [46–50]. During metastasis, cancer cells undergo dynamic metabolic shifts, such as increased glycolysis, altered lipid metabolism, and increased glutamine utilization, to meet the energetic and biosynthetic demands of invasion, survival, and colonization in distant organs [51,52]. Camkk2 plays crucial roles not only in PCa cell metastasis to and colonization of the lungs but also in the disruption of normal metabolic processes, such as glucose and lipid metabolism, leading to complications such as metabolic syndrome [51]. Additionally, the regulation of glutamine metabolism can upregulate ARPC1A in PCa cells, inducing cytoskeletal changes that promote the migration and invasion of tumor cells into the lungs [52]. Our research revealed that NEPC appears to exhibit unique metabolic characteristics. NEPC accumulates many depolarized mitochondria and relies on glycolysis for energy metabolism, resulting in increased lactate levels [53]. NEPC also specifically overexpresses the ADORA2A adenosine receptor. Mechanistically, ADORA2A enhances proline synthesis and increases the production of the intermediate metabolite NAD+ through the ERK/MYC/PYCR signaling axis, promoting SIRT6/7-mediated histone H3 deacetylation. Specific knockout of ADORA2A can reduce the occurrence of NEPC liver metastasis [54]. Enrichment analysis showed that genes related to metabolic pathways such as folate metabolism, selenium micronutrient network, fat digestion and absorption and cholesterol metabolism are enriched in liver metastases compared with metastases at other sites, potentially contributing to the initiation and progression of PCa liver metastasis [55]. Therefore, tumor cells likely need to adapt to new environments by altering their metabolic pathways, thereby facilitating the process of metastasis.
3. Liver Metastasis of PCa
In clinical diagnosis, early metastases to organs such as the liver, lungs, and brain often present with minimal or no symptoms. By the time of diagnosis, the disease has typically progressed to an advanced stage. In multiple phase III clinical trials, the initial proportion of lung metastases in mCSPC patients was greater than that of liver metastases. However, after chemotherapy, the proportion of lung metastases did not increase significantly, whereas the proportion of liver metastases increased, indicating that liver metastasis is closely associated with disease progression [39]. Aggressive PCa tends to metastasize to the liver, particularly among those who experience relapse after ARPI treatment [39]. Moreover, the survival time of mCRPC patients is related to the site of metastasis, with liver metastasis being associated with the worst prognosis and a median survival of only 13.5 months, followed by lung (19.4 months) and bone (21.3 months) metastases [56]. Therefore, further research into the molecular and cellular mechanisms of PCa liver metastasis holds promise for improving the survival of patients with advanced PCa and liver metastasis (Figure 1).
3.1 Neuroendocrine phenotype with a propensity for liver metastasis
Research indicates that patients with advanced PCa liver metastasis often exhibit AR-negative or neuroendocrine differentiation. In various cancers, the neuroendocrine phenotype is a significant source of liver metastatic tumors [57]. Compared with neuroendocrine tumor metastases at other sites, neuroendocrine tumor liver metastases are associated with a poorer prognosis. Therefore, exploration of how the neuroendocrine phenotype promotes tumor liver metastasis not only holds great significance for PCa treatment but also provides new research directions for liver metastasis in other neuroendocrine tumors.
Neuroendocrine tumors have the ability to produce neuron peptides such as serotonin (5-HT) [58]. Our recent research revealed that NEPC-secreted 5-HT is taken up by neutrophils via the serotonin transporter (SERT), which induces TGM2-mediated histone H3Q5 serotonylation. This process synergizes with PAD4-mediated histone H3 citrullination to promote NET release by neutrophils, thereby enhancing tumor cell liver metastasis. Inhibiting 5-HT uptake using the SERT inhibitor fluoxetine effectively reduces the occurrence of liver metastasis [59]. Neuroendocrine colon cancer (CRNE) is a common neuroendocrine phenotype that is often accompanied by liver metastasis in patients. Deng et al. systematically revealed significant differences in the cellular composition and microenvironment between primary lesions and liver metastases in CRNE liver metastasis (CRNELM) patients by integrating single-cell RNA sequencing and spatial transcriptomic sequencing. Pseudotime analysis revealed that pathways related to coagulation, hemostasis, and wound healing were significantly activated after tumor cells underwent epithelial–mesenchymal transition (EMT), suggesting that these pathways play critical roles in late-stage CRNELM [60]. Additionally, NEPC can be classified into two subtypes based on gene expression profiles: NE-classic and NE-liver. The NE-liver subtype exhibits a greater propensity for liver metastasis, with significant enrichment of immune response and complement pathways, indicating a unique immune microenvironment in the liver and the potential role of the complement pathway in NE tumor liver metastasis [33]. Our study revealed that thrombosis significantly promotes liver metastasis in NE-type tumors [61]. Specifically, NEPC expresses high levels of O-GalNAc catalyzed by the glycosyltransferase GALNT9. These glycosylation modifications are enriched primarily in ANXA2 and induce platelet activation and thrombosis by binding to and activating the MBL coagulation pathway, thereby promoting NEPC liver metastasis. Targeting GALNT9 or O-GalNAc glycosylation effectively inhibits liver metastasis in NE tumors [61]. In summary, thrombosis induced by the coagulation cascade is a crucial mechanism for NE tumor liver metastasis, providing potential directions for developing targeted therapies. In summary, NE tumors appear to promote liver metastasis through multiple mechanisms. At the molecular level, activation of the coagulation cascade to promote thrombosis and colonization in the liver is a shared characteristic of NE tumor liver metastasis. Additionally, patients with NE tumors often experience neuroendocrine system dysregulation. Whether NEPC secretes neuroendocrine substances other than 5-HT requires further investigation.
3.2 The role of the liver immune microenvironment in metastasis
The liver is enriched with various immune cells, including NKT cells, NK cells, Kupffer cells, neutrophils, T cells, and DCs, which collectively maintain the balance between clearing foreign antigens and preventing excessive immune responses that could cause tissue damage, thereby shaping a unique immune microenvironment in the liver [62]. Therefore, understanding the specialized microenvironment formed by these cells in the liver can better explain the propensity of tumor cells for liver metastasis.
Unlike those in other organs, many immune cells in the liver exhibit tissue-resident phenotypes. The most common tissue-resident immune cells include Kupffer cells, NKT cells, NK cells and ILC1 cells. Kupffer cells, the most frequently studied type, are a unique type of innate immune cell in the liver that play crucial roles in various liver diseases, homeostasis maintenance, injury repair, and tumorigenesis [63]. Kupffer cells can phagocytose tumor cells that metastasize to the liver, exerting antitumor effects. The ERMAP/Gal-9/dectin-2 axis, constituting an antibody-independent nonclassical "eat me" signal, specifically promotes Kupffer cell-mediated phagocytosis of tumor cells, thereby inhibiting liver metastasis. Activating the ERMAP/Gal-9/dectin-2 signaling axis could serve as a strategy to enhance Kupffer cell phagocytosis and antitumor innate immunity, offering potential clinical strategies for treating and preventing liver metastasis [64]. β-Glucan activates Kupffer cells, inhibits cancer cell proliferation, and elicits productive T-cell-mediated antimetastatic responses in pancreatic cancer mouse models [65]. Thus, Kupffer cells are the first line of defense against tumor liver metastasis. However, TAMs are known to promote tumor growth in many cancers, and whether Kupffer cells have similar protumor mechanisms remains to be further studied. NKT cells, NK cells, and ILC1 cells are generally considered to have strong tumor-killing capabilities and can reside in the liver, maintaining the capacity for self-renewal and differing from peripheral circulating cells, thereby forming a unique phenotype in the liver [66–68]. Compared with other organs, NKT cells are more enriched in the liver, where they perform unique functions [66,69]. The gut microbiota can influence the balance of primary and secondary bile acids, affecting CXCL16 secretion by liver sinusoidal endothelial cells and regulating NKT cell aggregation, which is crucial for NKT cell antitumor functions [70]. However, certain NKT cell subtypes, such as iNKT17 cells, have been shown to release IL-22, which acts on endothelial cells to induce endothelial aminopeptidase N, increasing endothelial permeability and promoting tumor cell extravasation [71]. Under steady-state conditions, the liver contains conventional NK cells (cNK cells), defined as CD49a-CD49b+ cells, and tissue-resident CD49a+CD49b- type 1 innate lymphoid cells (ILC1 cells) [72,73]. NK cells are cytotoxic lymphocytes that play a significant protective role in tumor development and metastasis [74,75]. The size of the NK cell pool in the liver itself can determine metastatic growth. In preclinical models, maintaining NK cell abundance through IL-15-based adjuvant immunotherapy successfully prevented liver metastasis and prolonged survival [76]. NK cells can also eliminate hepatic stellate cells and inhibit hepatocyte proliferation to inhibit liver fibrosis, thereby reducing the occurrence of tumor liver metastasis [77–79]. Compared with those in primary lesions, T cells, NK cells, and NKT cells in CRNELM exhibit a stressed-like phenotype, with reduced cytotoxicity scores and an immunosuppressive state, indicating severe functional impairment [60]. In fact, a recent study by Ducimetière et al. revealed that NK cells have durable antimetastatic functions in MC38-injected mice, whereas tissue-resident ILC1 cells are essential for inhibiting CRC liver metastasis in the early stages [80]. The microenvironment of colorectal cancer liver metastases results in the formation of a CD49a+ NK cell subset with ILC1 cell-like characteristics, and these cells exhibit the strongest cytotoxicity among infiltrating NK cells. CXCR3 drives NK cell accumulation in CXCL9- and CXCL10-enriched regions, promoting their colocalization with macrophages. Macrophages regulate NK cell plasticity through dual mechanisms of CXCL9 chemotaxis and TGF-β induction, modulating their antitumor functions [81]. Tissue-resident ILC1 cells in the liver specifically express GPR34, and tumor-derived lipid metabolites such as lysophosphatidylserine can inhibit ILC1 cell antitumor activity via GPR34. Antagonizing GPR34 induces potent ILC1 cell-mediated antitumor immunity, suppressing the growth of liver cancer, colorectal cancer, and other solid tumors [82]. However, the mechanisms by which tumor cells, especially PCa cells, evade the killing effects of liver-resident immune cells remain to be elucidated. In conclusion, diverse immune cells in the liver, including Kupffer cells, NKT cells, NK cells, and ILC1 cells, play complex and critical roles in tumor liver metastasis. These cells participate in antitumor immune responses through mechanisms such as phagocytosis, cytotoxicity, and immune regulation, but they can also be exploited by tumor cells to promote colonization and metastasis.
3.3 The role of non-immune cells in liver metastasis
Liver injury and fibrosis create a favorable microenvironment for tumor metastasis through multiple mechanisms. Clinical evidence shows that colorectal cancer patients with liver fibrosis have a higher risk of developing liver metastasis compared to those without fibrosis [83]. Mechanistically, liver damage promotes excessive extracellular matrix (ECM) deposition, and activated hepatic stellate cells produce abundant fibronectin. These changes not only alter liver architecture but also recruit bone marrow–derived immune cells, collectively establishing a niche that supports tumor cell colonization and growth [84,85]. In PCa, systemic therapies such as hormonal therapies and chemotherapy may unintentionally promote liver metastasis by inducing hepatotoxicity [86,87]. ARPIs like bicalutamide and abiraterone are associated with varying degrees of liver damage [88,89]; abiraterone, for instance, causes elevated liver enzymes in ~6% of patients [89]. Therapy-induced hepatic injury may foster metastasis via paracrine signaling, stellate cell activation, fibrotic and inflammatory feedback loops, ECM remodeling, and immune modulation [90]. Notably, under conditions of liver injury, activated stellate cells can suppress NK cell proliferation, thereby impairing NK-mediated maintenance of tumor dormancy and ultimately facilitating tumor awaken [76]. These findings suggest that ARPI-induced liver injury may establish a pre-metastatic niche while simultaneously activating stellate cells to reactivate dormant tumor cells. At the molecular level, the hepatocytes express AR in both the cytoplasm and nucleus [91]. Androgens regulate hepatic expression of key cytokines such as TGF-β and VEGF [92]. Notably, TGF-β, a central mediator of liver fibrosis, is also highly upregulated in PCa liver metastases, where it facilitates invasion, immunosuppression, and supportive stroma formation [93–95]. E-cadherin regulation is another critical factor during PCa liver metastasis. Downregulation of E-cadherin at the primary site enables EMT and cell migration. However, within the hepatic microenvironment, hepatocytes may suppress EGFR signaling to induce mesenchymal–epithelial transition, restoring E-cadherin expression to promote metastatic colonization [96,97]. Subclones lacking E-cadherin undergo apoptosis, while those re-expressing it survive through downstream kinase activation [98]. These findings highlight the need for management of treatment-related hepatotoxicity and support the development of combination strategies targeting liver fibrosis and pre-metastatic niche.
4. PCa Nonbone Metastasis Models
Compared with other cancers, such as colon cancer, lung cancer, pancreatic cancer, and melanoma, which are prone to nonbone metastasis, research on the cellular and molecular mechanisms of nonbone metastasis in PCa remains limited. This is largely due to the lack of well-established models for studying nonbone metastasis in PCa. Initially, researchers induced oncogene expression in the mouse prostate to promote PCa formation and metastasis. The TRAMP model, which utilizes high SV40 T antigen expression in the prostate, successfully induces PCa development [99,100]. TRAMP mice effectively mimic the natural progression of PCa and can be used to study different disease stages, which correlate with the age of the mice. TRAMP mice develop prostatic intraepithelial neoplasia (PIN) at approximately 12 weeks, which progresses to adenocarcinoma. By 28–32 weeks, TRAMP mice typically develop advanced disease, leading to lymph node and distant metastases [99,100]. With advances in gene-editing technology, transgenic mice expressing Cre recombinase under the probasin (Pb) promoter have provided a foundation for generating other genetically engineered mouse models (GEMMs) with prostate epithelial-specific gene deletions. The Pten conditional knockout model recapitulates human disease progression, starting with PIN and advancing to invasive adenocarcinoma and subsequent metastasis [101]. Homozygous Pten knockout successfully induces PCa and lung metastasis, although the number of metastases is limited [101]. This was the first PCa mouse model in which metastasis was induced by the knockout of an endogenous gene. To increase tumorigenicity and malignancy, researchers began developing double-gene knockout models, such as PbCre+PtenΔ/ΔP53Δ/Δ, PbCre+Rb1Δ/ΔP53Δ/Δ, and PbCre+PtenΔ/ΔRb1Δ/Δ [35,36,102,103]. Similar to PbCre+PtenΔ/Δ, PbCre+PtenΔ/ΔP53Δ/Δ primarily shows occasional lung metastasis and more frequent lymph node metastasis [102]. PbCre+Rb1Δ/ΔP53Δ/Δ and PbCre+PtenΔ/ΔRb1Δ/Δ exhibit more distant metastases to organs such as the liver, lungs, and lymph nodes [35,36]. The triple-knockout PbCre+PtenΔ/ΔRb1Δ/ΔP53Δ/Δ model displays a more aggressive phenotype, with a median survival of only 16 weeks and rapid metastasis to the liver, lungs, and bones, making it a suitable model for studying spontaneous PCa with liver and lung metastasis [35]. In addition to GEMM-derived tumor models, the RapidCaP method involves surgical injection of lentiviral Cre-luciferase (LV-Cre/Luci) into the prostate of Ptenloxp/loxp mice, leading to Pten and Trp53 deletion and triggering mPCa. High rates of metastasis to the lymph nodes, spleen, and liver are observed within months [104].
Although GEMMs better mimic disease progression, metastasis remains inconsistent, highly variable among individuals, and time-consuming to develop, requiring extensive breeding of multiple genetically modified mouse lines. Therefore, there is an urgent need for rapid and stable models to support research on nonbone metastasis in PCa. Current animal models of tumor metastasis can be established through various methods, including intracardiac injection [105], tail vein injection [71], metastatic site inoculation [106], and spleen implantation [98]. In PCa, intracardiac [107], intrasplenic [98], or hemisplenic injections [108] can induce metastasis to the liver, lungs, and spleen. While these methods effectively establish metastatic models, they do not fully replicate the process of tumor cell dissemination from the primary prostate site in the context of nonbone metastasis. To address this, our team developed a novel metastasis model by orthotopically injecting PCa -derived organoids from PbCre+Rb1Δ/ΔP53Δ/Δ mice into immunocompetent mice, generating a stable, rapid, and highly efficient liver metastatic PCa model [109]. This method is not only efficient but also suitable for studying interactions between PCa cells and immune cells.
In vitro studies rely primarily on PCa cell lines, patient-derived organoids (PDOs), and patient-derived xenografts (PDXs) to investigate metastatic features in clinical PCa patients [110]. Currently, most PCa cell lines are derived from metastatic tumor tissues. However, among these, only DU145 cells originate from brain metastases, whereas others are derived primarily from lymph node metastases (e.g., LNCaP, NCI-H660, ALVA-55, and LAPC-4) and bone metastases (e.g., PC-3, ALVA-41, ALVA-101, MDA PCa 2a, and MDA PCa 2b) [110]. Cell lines derived from liver, lung, or other solid-organ metastases are lacking. PDOs also serve as valuable in vitro models, retaining the molecular features observed in patient tumors [111,112]. Studies have shown that approximately 70% of soft tissue tumor samples and 30% of bone tumor samples can generate organoids stably cultured for 1–2 months, but stable organoids passaged for more than six months are derived mostly from bone and lymph node metastases [111]. The lack of PDOs from solid-organ metastases is partly due to current culture methods favoring normal epithelial cell growth, making isolation of tumor cells from liver and lung metastases challenging. PDX models involve direct transplantation of freshly isolated tumor tissues, such as primary or metastatic lesions, from patients into immunodeficient mice. PDX models preserve the unique mutational characteristics and heterogeneity of the original tumor and hold promise for personalized treatment. PDX models can also be used to study PCa metastasis in mice. A method involving subrenal capsule transplantation in SCID mice combined with androgen level modulation achieves >95% tumor formation rates and induces metastasis to the liver, lungs, kidneys, and spleen [113]. While this method can generate nonbone metastases and retain patient tumor characteristics, the lack of immune and stromal cell interactions in SCID mice limits the ability of this model to fully replicate the in vivo microenvironment. Therefore, the use of humanized mice may better simulate the mechanisms of nonbone metastasis in PCa.
5. Diagnosis and Treatment of mPCa
5.1 Diagnosis of mPCa
In the study of nonbone metastasis in PCa, ultrasound, CT, MRI, and PET are the most commonly used imaging modalities. 68Ga- or 18F-labeled PSMA PET has shown promising results in detecting distant metastasis of PCa [114–118]. In the detection of liver metastasis, 68Ga-labeled PSMA PET may have an advantage over 18F-labeled PSMA PET, as 18F-PSMA tends to be excreted through the liver, leading to increased physiological uptake that can compromise detection accuracy [119]. However, a significant proportion of metastatic lesions (22.3%) lack PSMA expression, as PSMA expression is indirectly regulated by AR, and PCa with nonbone metastasis are often AR-negative [120–122]. Therefore, PSMA PET still has the potential for missed diagnoses. 18F-FDG PET-CT is widely used in various cancers, but its application in PCa is limited due to the low glucose metabolism of PCa cells [123]. However, PCa with nonbone metastasis, especially visceral metastatic NEPC, often exhibit high uptake on FDG-PET, suggesting unique glycolytic characteristics, which may be related to microenvironmental reprogramming in metabolically active organs such as the liver [124]. In tumors with low AR and PSMA expression, changes in genes related to 18F-FDG uptake are positively correlated with increased glucose uptake [125]. This evidence suggests that 18F-FDG PET-CT may perform better than PSMA PET-CT in detecting tumor lesions with low PSMA expression. Recently, Ahumada et al. conducted a paired clinical study using three imaging agents—18F-FDG for glucose metabolism, PSMA, and somatostatin receptor (SSTR) for neuroendocrine tumors—and found that among 273 lesions in 8 NEPC patients, PSMA was positive in only 174 lesions, SSTR was positive in only 59 lesions, and 18F-FDG was detected the most, in 182 lesions [126]. The false-negative rate of PSMA PET-CT for liver lesions was as high as 20%, with most of these lesions being positive for 18F-FDG. Therefore, PET imaging with the above radiotracers provides some supportive value in the diagnosis of NEPC. PSMA PET has limited diagnostic performance for nonbone metastases in PCa, especially in NEPC. DLL3 is expressed on the surface of small cell lung cancer and NEPC cells. A PET tracer has been developed using a DLL3-targeted antibody SC16 labeled with 89Zr (89Zr-DFO-SC16), enabling noninvasive identification of DLL3-positive NEPC lesions [127]. In a phase 1/2 clinical trial (NCT04199741), 89Zr-DFO-SC16 PET-CT has been demonstrated to be safe and feasible [128]. Thus 89Zr-DFO-SC16 PET-CT holds promise as a NEPC-specific imaging. A multimodal imaging approach holds promise for improving the accuracy of nonbone metastasis detection. Taranda et al. used serial two-photon tomography (STPT) combined with traditional histology in a genetically engineered RapidCaP mouse model to characterize PCa and its metastasis. STPT can detect single tumor-initiating cells throughout the prostate, and subsequent IF analysis revealed the transition from normal to transformed epithelial tissue and the escape of cells from tumor foci. STPT imaging of the liver and brain revealed the distribution of multiple metastatic foci in the liver and the invasion of early metastatic cells in the brain. This imaging and data analysis pipeline provides a highly versatile whole-organ platform for studying the mechanisms of cancer initiation and progression [129]. Together, there is an urgent clinical need to study the molecular mechanisms of PCa progression and metastasis, explore noninvasive and effective diagnostic and therapeutic strategies, and develop new methods and technologies for the effective treatment and evaluation of mPCa, which is of great clinical importance for timely intervention and prolonged survival. PCa metastases often involve multiple organs throughout the body, and their treatment focuses on systemic therapy, making highly sensitive and specific diagnostic imaging modalities for systemic mPCa of great clinical value (Table 1).
When the imaging diagnosis is uncertain, image-guided biopsy can be used to confirm the presence of metastasis. Biopsy of metastatic lesions can help determine the histological type of metastatic cancer cells, such as adenocarcinoma or NEPC. Therefore, biopsy remains the gold standard for diagnosing PCa metastases and provides information for selecting the best treatment strategy. For example, liver metastases originating from PCa can be identified by positive AMACR, negative cytokeratin 7, and negative cytokeratin 20 expression [149]. However, since mCRPC patients are in the advanced stages of the disease, they often refuse biopsy, so diagnosis requires the integration of multiple sets of clinical information and the development of more specific and sensitive, less invasive diagnostic methods.
In recent years, liquid biopsy, as a representative in vitro tumor diagnostic and monitoring method, has gradually shown advantages. For example, peripheral blood CHGA, CEA, and NSE levels can be used as alternative markers for diagnosing NEPC [16,130–132]. Circulating tumor DNA (ctDNA) is an emerging biomarker for a range of solid malignancies, including mCRPC [133]. ctDNA analysis is theoretically applicable to any patient who can provide a blood sample and can be performed repeatedly on the same patient with minimal invasiveness and moderate cost. Cell-free DNA (cfDNA) is shed into the blood by both nonmalignant and cancer cells [134,135]. In mCRPC patients, the proportion of tumor-derived cfDNA often exceeds 1%, making it possible to detect tumor genomic profiles from cfDNA [136–139]. Mutations and copy number changes in plasma cfDNA from mCRPC patients are consistent with the somatic landscape established through metastatic tissue profiling [22,136,138–143]. W. Wyatt et al. collected 45 matched metastatic tissues and liquid biopsies from mCRPC patients and quantified the concordance of somatic changes in key driver genes [144]. ctDNA detection is sufficient to identify all driver DNA alterations present in matched metastatic tissues and supports the development of DNA biomarkers to guide the management of mCRPC patients based solely on ctDNA [144,145]. The concordance between cfDNA and biopsy tissue genomic changes is significantly greater in NEPC patients than in CRPC patients, indicating that cfDNA has promise as an effective biomarker for diagnosing NEPC [146]. Evaluation of the methylation-based NEMO panel in cfDNA provides a scoring system for the NEPC phenotype and can be used for the noninvasive detection of pathologically confirmed NEPC [147]. Genomic sequencing of cfDNA can also aid in identifying NEPC subtypes that are sensitive to platinum-based therapies [130,148]. Moreover, the methylation patterns of cfDNA can be used to characterize tumor-associated epigenetic changes [146]. Therefore, liquid biopsy is expected to serve as a less invasive detection method for disease progression in the future.
5.2 Treatment of mPCa
In recent years, treatment strategies for mPCa have undergone significant changes, transitioning from traditional ADT to multidrug combination therapies, especially in cases of nonbone metastasis, where the complexity and heterogeneity of treatment are greatly increased [150,151]. ADT typically involves reducing testosterone production through surgical or medical castration, achieved via orchiectomy or gonadotropin-releasing hormone agonists or antagonists. For patients with mCSPC, current treatments advocate early combination therapy to delay resistance progression and maximize overall survival (OS) [150,152–158]. Several phase III trials, such as CHAARTED [159], STAMPEDE [160], and LATITUDE [89], have consistently shown that combining ADT with docetaxel and novel ARPIs such as abiraterone, enzalutamide, and apalutamide can prolong progression-free survival (PFS) and OS in mCSPC patients. Additionally, the PEACE-1 [161] and ARASENS [162] studies have advanced triple therapy (ADT + docetaxel + ARPI) into clinical practice, demonstrating significant survival benefits in mCSPC patients with high-volume disease. In the ARASENS study, the median OS of patients with visceral metastasis who received triple therapy improved by 7 months compared with that of patients who received only docetaxel treatment (49.0 months vs. 42.0 months) [162]. In addition to using ARPIs, therapies such as RIPTAC and PROTAC-mediated AR degradation are also under exploration [163,164]. Taken together, some mPCa tumors retain AR pathway dependence, and AR-directed therapy remains of clinical relevance. However, existing evidence has not sufficiently differentiated the efficacy of this approach in cases of nonbone metastasis versus bone metastasis, suggesting that individualized treatment based on burden and organ specificity is needed in clinical practice (Table 2).
When the disease progresses to the mCRPC stage, treatment strategies increasingly rely on precise stratification based on tumor molecular characteristics and metastatic sites. Docetaxel remains the foundational first-line treatment, and for patients with lung or liver metastasis, ARPI treatment still provides clear survival benefits, although its efficacy varies among subgroups [165,166,185,186]. Data from studies such as AFFIRM and PREVAIL show that the median OS for patients with lung metastasis treated with enzalutamide is superior to that of patients with liver metastasis (e.g., in the PREVAIL trial: 32.4 months for lung metastasis vs. 18.9 months for liver metastasis), indicating a significantly worse prognosis and limited response to current treatments in patients with liver metastasis [165–167]. Further research revealed that liver metastasis in PCa is often associated with decreased AR expression, neuroendocrine transdifferentiation, and high genomic instability, which are closely related to ARPI treatment resistance [39]. NEPC is typically treated with chemotherapy regimens derived from those used for small cell lung cancer, primarily consisting of a combination of cisplatin and etoposide [15,131,148]. AURKA is highly expressed in NEPC, and using the AURKA inhibitor alisertib can improve patient prognosis [176]. In addition, EZH2 inhibitors are also potential therapeutic targets [179,180]. Although conventional immune checkpoint inhibitor therapies have shown limited benefit in PCa, novel approaches such as T-cell engagers, vaccines, and CAR-T therapies still represent meaningful research directions for mPCa [184].
For patients with liver metastasis, current standard treatments include chemotherapy, ARPIs, and radioligand therapy, but overall efficacy remains poor [39]. Although radionuclide therapy (e.g., the VISION study) has improved OS in the overall mCRPC population, its efficacy is limited in patients with low PSMA expression and neuroendocrine phenotypes, necessitating further evaluation [168,187]. As a result, some studies are exploring immunotherapy combination strategies or novel radioligands targeting PSMA-negative markers (e.g., FAP-PET) to improve treatment outcomes, although these are still in the exploratory stage [169]. NEPC is more prone to liver metastasis, making NEPC-specific targets promising for targeted therapy. DLL3 is highly expressed in NEPC, and treatment of DLL3-expressing NEPC with the antibody-drug conjugate SC16LD6.5 has shown effective responses, suggesting DLL3 as a potential therapeutic target for NEPC [175]. In addition, the development of liver metastasis often accompanies tumor clonal evolution induced by treatment selection pressure, with some tumor cells potentially evading traditional treatments by acquiring neuroendocrine phenotypes or enhancing DNA damage response pathway activity [32,188]. As mentioned in Part 2, DDR gene mutations are enriched in mPCa, and PARPs play a key role in DNA repair. Phase III trials such as PROPEL [181], MAGNITUDE [182], and TALAPRO-2 [183] have shown that patients with mCRPC derive significant radiographic progression-free survival benefits from the combination of PARP inhibitors and ARPIs. These findings suggest that the use of PARP inhibitors or combination therapies targeting DDR-related pathways may hold promise in these patients [170,171]. The immune microenvironment of liver metastasis is highly immunosuppressive, with mechanisms such as TGF-β signaling, MDSC recruitment, and T-cell exhaustion rendering immunotherapy ineffective [39]. Thus, liver-directed radiotherapy or TGF-β inhibitors may help alleviate immune evasion and enhance the efficacy of systemic immunotherapy [172–174].
Notably, while lung metastasis in mCRPC is associated with a worse prognosis than bone metastasis, it is still better than that associated with liver metastasis. In studies such as PREVAIL, lung metastasis patients responded relatively well to ARPIs, indicating that the biological characteristics of their tumors are more similar to those of classic AR-driven CRPC [165,166]. For those with NEPC or AR-negative tumors, platinum-based chemotherapy (e.g., carboplatin + etoposide) or participation in clinical trials exploring novel immunotherapy combination strategies is recommended [155,189]. However, there is currently no established standard treatment for DNPC patients. Our team has identified Gremlin1 as an important therapeutic target in advanced PCa, with particularly high expression in DNPC. Gremlin1 promotes PCa progression and androgen deprivation resistance by directly binding to FGFR1 and activating the FGFR1/MAPK signaling pathway, which is closely associated with FGF signaling activation. We have developed monoclonal antibodies against human or murine Gremlin1 and demonstrated their efficacy and safety in mouse models. Therefore, under a suitable dosing window, the use of Gremlin1 antibodies in patients may avoid unnecessary side effects and represent a novel approach for DNPC treatment [177]. As a key pathway in DNPC, the FGFR signaling pathway represents a promising therapeutic target [18]. Thus, future strategies targeting FGFR1 and GREM1 hold potential for the treatment of DNPC patients [178].
In summary, current treatments for nonbone mPCa, especially PCa with lung and liver metastasis, face significant challenges. Patients with lung metastasis have a relatively favorable prognosis with existing ARPI therapies and are recommended to follow standard CRPC treatment pathways, whereas patients with liver metastasis should be considered a high-risk subtype and treated with multimodal combination strategies and novel targeted interventions. Future efforts could focus on establishing liver metastasis-specific biomarker screening systems, developing more precise imaging and liquid biopsy methods, and conducting multicenter clinical trials to validate personalized treatment plans, ultimately improving the prognosis of these patients.
6. Conclusion and Perspective
Cancer metastasis is the leading cause of patient death, yet the mechanisms underlying metastasis and effective interventions remain poorly understood [68,190–192]. PCa is one of the most common malignancies in men, and its metastatic mechanisms and clinical treatment strategies have long been key areas of research. Although bone metastasis is relatively common in PCa patients, patients who experience recurrence after treatment often present with nonbone metastases, which pose a serious threat to their survival.
The mechanisms of nonbone metastasis in PCa involve multiple factors, including genomic mutations, PCa phenotypes, microenvironmental regulation, and metastatic organ-specific microenvironments. Common genomic alterations in PCa include TMPRSS2-ERG fusion, AR amplification, gain-of-function mutations in FOXA1, and loss-of-function mutations in SPOP [4]. Moreover, mutations in DNA damage repair genes are frequently observed in mPCa, particularly in brain metastatic PCa. Notably, the loss of PTEN in mPCa is considered a major driver of metastasis, followed by the loss of RB1 and TP53, as well as the double loss of RB1 and TP53, resulting in the NEPC phenotype.
During the progression of PCa, lineage plasticity leads to the emergence of various subtypes. NEPC, a highly aggressive subtype of PCa, exhibits strong metastatic capabilities, particularly a significantly increased incidence of liver metastasis [15,20]. The development and progression of NEPC are unclear and involve lineage plasticity acquisition and the expression of neuroendocrine markers. Studies suggest that NEPC may have unique metabolic pathways, with its specific metabolites and secreted neuroendocrine factors potentially serving as therapeutic targets. Furthermore, the tendency of NEPC to metastasize to the liver provides important insights for research on other neuroendocrine tumors. DNPC represents another aggressive phenotypic variant of PCa. However, its diagnosis relies primarily on exclusion methods, as it is negative for both AR and NE markers. There is a lack of definitive therapeutic targets for DNPC. Future research could integrate multiomic data, including genomic, transcriptomic, proteomic, and metabolomic data, to construct molecular profiles of DNPC and identify specific biomarkers and potential therapeutic targets.
Different types of cancer exhibit distinct organotropism. Differences in organ-specific microenvironments are key determinants of this organotropism. This selective metastasis is regulated by multiple mechanisms, including chemokines, cell–cell interactions, and immune modulation, which collectively guide tumor cells to migrate, colonize, and grow in specific organs. The bone is the most common site of metastasis in PCa. The bone microenvironment contains unique components, such as osteoclasts, osteoblasts, osteocytes, mineralized bone matrix, and abundant cytokines that together create a “fertile soil” supporting tumor growth [193–195]. Furthermore, the bone marrow serves as a critical niche for dormant tumor cells, where factors such as Gas6, BMP4, and BMP7 regulate the homing and dormancy of PCa cells [196].
In contrast to the well-studied PCa bone metastases, the mechanisms of PCa nonbone metastases remain incompletely understood. The liver is rich in immune cells and maintains an “immune-tolerant” microenvironment to balance immune surveillance and tolerance to external antigens [62]. This suppressive environment facilitates tumor immune evasion. Although circulating tumor cells can enter the liver parenchyma via sinusoidal vessels, they often encounter initial growth suppression and may remain dormant for a long time [11,197,198]. This dormancy is at least partly mediated by immune regulation, such as the secretion of IFN-γ by liver-resident NK cells, which is essential for maintaining dormancy of breast cancer disseminated tumor cells [76]. Studies have shown that chemotherapy may induce the awaken of dormant tumor cells [199]. In PCa patients receiving ARPIs, treatment-associated liver injury and fibrosis may stimulate the awakening of dormant cancer cells in the liver, potentially triggering recurrent liver metastases. The lung, with its extensive vasculature and large capillary surface area, provides a favorable site for circulating tumor cells arrest, extravasation, and colonization. Moreover, tissue-resident macrophages in the lung, such as alveolar and interstitial macrophages, play key roles in regulating metastasis [200,201]. In summary, organ-specific metastasis in PCa reflects distinct organotropism, which is shaped by multiple interactions within each organ’s microenvironment. A deeper understanding of how different organ microenvironments regulate tumor cell fate may offer new therapeutic directions to prevent and manage PCa metastases.
Among all nonbone metastatic sites, metastasis to the liver is associated with the worst prognosis, making understanding liver metastasis in PCa crucial for improving patient outcomes. Here, we elaborate on the liver metastasis of PCa and provide biologically significant insights. Patients with PCa liver metastasis typically present with advanced-stage disease. Notably, ARPI-induced liver injury may potentially facilitate liver metastasis progression. Under liver injury conditions, activated hepatic stellate cells can further reactivate dormant tumor cells. These mechanisms provide important insights for understanding liver metastasis in PCa, particularly in the context of treatment-induced NEPC. Moreover, PCa as a typical immunosuppressive "cold tumor", evades elimination by various resident immune cells in the liver through mechanisms that remain unclear. In the future, research on resident immune cells in the liver may explain the specificity of liver metastasis in PCa and lead to the development of new targeted therapeutic strategies.
The role of the nervous system in PCa has gained increasing attention in recent years [202]. Nerves and neural factors have potential clinical applications as biomarkers for patient stratification. However, in other cancer types, the roles of different nerves, including sympathetic, parasympathetic, and sensory nerves, are not entirely consistent [203]. Whether these nerves exert protumor or antitumor effects in PCa remains to be elucidated. Moreover, the nervous and immune systems share extensive crosstalk in signal transduction, with neurotransmitters directly modulating immune cell function by binding to their cognate receptors on immune cells [204]. Therefore, investigating the differential immunomodulatory effects of distinct neural subtypes in PCa represents a significant research frontier. Beyond the regulation of tumor metastasis by nerves at the primary site, it remains unclear whether nerves at the nonbone metastatic site further promote tumor growth.
Although nonbone metastatic models of PCa have been partially established, significant areas for improvement remain. First, while bone metastasis is more prevalent in PCa patients, mouse models predominantly develop nonbone metastases, a discrepancy largely attributed to interspecies differences. Consequently, findings from murine studies may not always translate effectively to patients. Future research could prioritize clinically relevant phenomena observed in patients before experimental validation in models. Second, existing murine nonbone metastasis models lack heterogeneity. For example, liver metastasis studies often rely on NEPC models while ignoring non-NEPC subtypes. Only through stable establishment of representative nonbone metastatic models can the underlying mechanisms be thoroughly investigated.
The diagnosis and treatment of nonbone mPCa face significant challenges due to disease heterogeneity. Imaging techniques, including PSMA PET and 18F-FDG PET-CT, are crucial for detecting metastases, although PSMA PET has limitations in detecting AR-negative tumors. Emerging liquid biopsy technologies now enable noninvasive profiling of tumor subtypes such as NEPC and mutational signatures through ctDNA analysis, facilitating real-time monitoring of disease progression and therapeutic response. In terms of treatment, there is still a lack of highly specific and effective therapeutic approaches. In the future, developing more precise diagnostic methods, defining optimal treatments for different PCa mutations and subtypes, and investigating novel targeted therapeutic strategies are expected to further improve patient prognosis.
Declarations
Ethics Statement
Not applicable
Consent for Publication
Not applicable
Availability of Data and Material
Not applicable
Funding
The work was funded by grants from the National Natural Science Foundation of China NSFC-U23A20454, and NSFC82372873, Shanghai Pilot Program for Basic Research-Shanghai Jiao Tong University (21TQ1400225), the Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20181706), the Innovative research team of high-level local universities in Shanghai and the 111 project (B21024), and Collaborative Innovation Center for Clinical and Translational Science by Ministry of Edition & Shanghai (CCTS-202402), and RJZH25-005 from Renji Hospital to H.H.Z. This work was also supported by the Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No. 2024ZD0536300), the China Postdoctoral Science Foundation (Grant No. GZC20241049) and the Shanghai Natural Science Foundation (Grant No. 24ZR1444500).
Competing Interests
Helen He Zhu is a member of the Editorial Board of the journal Cancer Heterogeneity and Plasticity. The author was not involved in the journal’s review of or decisions related to this manuscript. The author has declared that no other competing interests exist.
Author Contributions
H.H.Z, N.J, and G.Y.D drafted the manuscript. P.H.X assisted in creating cartoon elements in Graphical drawing. All authors reviewed and approved the final manuscript.
Abbreviations
The following abbreviations are used in this manuscript:
- PCa:
- prostate cancer
- mPCa:
- metastatic prostate cancer
- ADT:
- androgen deprivation therapy
- ARPI:
- androgen receptor pathway inhibitor
- CSPC:
- castration-sensitive prostate cancer
- CRPC:
- castration-resistant prostate cancer
- mCRPC:
- metastatic castration-resistant prostate cancer
- mCSPC:
- metastatic castration-sensitive prostate cancer
- NEPC:
- neuroendocrine prostate cancer
- DNPC:
- double-negative prostate cancer
- DDR:
- DNA damage repair
- HRD:
- homologous recombination deficiency
- CRNE:
- neuroendocrine colon cancer
- CRNELM:
- neuroendocrine colon cancer liver metastasis
- EMT:
- epithelial–mesenchymal transition
- GEMM:
- genetically engineered mouse model
- PDO:
- patient-derived organoid
- PDX:
- patient-derived xenograft
- ctDNA:
- circulating tumor DNA
- cfDNA:
- cell-free DNA
Table 1 Diagnostic Strategies for mPCa
| Category |
Modality |
Principle |
Key Findings/Limitations |
Implication for Mets |
| Imaging |
68Ga-/18F-PSMA PET-CT |
PSMA targeting via AR regulation |
Good for bone and soft tissue; 18F excreted via liver → false positives; ~22% lesions PSMA-negative [114–122] |
Limited sensitivity in liver metastasis and AR-negative NEPC; may miss lesions |
| Imaging |
18F-FDG PET-CT |
Glucose metabolism |
Limited in classic PCa due to low glycolysis; high uptake in NEPC and liver metastases [123–126] |
May outperform PSMA PET in AR-low, PSMA-negative lesions, esp. NEPC |
| Imaging (Preclinical) |
89Zr-DFO-SC16 |
DLL3 target |
DLL3 is specifically expressed in NEPC, serves as a target for NEPC imaging [127,128] |
May useful for detecting NEPC with nonbone metastasis |
| Imaging (Preclinical) |
STPT (Serial Two-Photon Tomography) |
Whole-organ 3D imaging in GEMMs |
Visualizes early escape, liver/brain micromets in mouse models [129] |
Preclinical only; informs spatial metastasis evolution |
| Tissue Biopsy |
Biopsy of metastases |
Histopathologic confirmation |
Gold standard for diagnosis; limited in advanced mCRPC due to patient condition |
Useful for subtyping (e.g., AMACR+, CK7–, CK20– for liver mets) [4] |
| Liquid Biopsy |
ctDNA/cfDNA, Serum markers (CHGA, CEA, NSE) |
Tumor DNA from plasma/ Protein markers for NEPC |
High concordance with metastasis tissue; captures AR/NE drivers and epigenetic patterns [16,22,130–148] |
Non-invasive, useful in advanced mCRPC; applicable to NEPC subtyping |
This table summarizes current imaging and molecular diagnostic tools used in identifying nonbone mPCa. Special emphasis is placed on the strengths and limitations of each modality in detecting visceral metastases (particularly liver and NEPC), highlighting the clinical value of multimodal imaging and liquid biopsy in mCRPC staging and subtype classification.
Table 2 Treatment Strategies for mPCa.
| Disease Stage |
Main Treatments |
Key Evidence |
Outcomes/Challenges |
Future Directions |
| mCSPC |
Triple therapy:ADT + Docetaxel or ARPI (abiraterone, enzalutamide, apalutamide) |
CHAARTED [159], STAMPEDE [160], LATITUDE [89], PEACE-1 [161], ARASENS [162] |
Prolonged PFS/OS; triple therapy improves OS in visceral metastases (e.g., ARASENS: 49.0 vs. 42.0 months) |
Stratified treatment by metastatic burden and organ type |
| mCRPC-Lung metastasis |
Docetaxel +ARPIs (e.g., enzalutamide) |
AFFIRM [165], PREVAIL [166,167] |
Better prognosis vs. liver metastasis (e.g., PREVAIL: 32.4 vs. 18.9 months); relatively preserved AR signaling |
Standard CRPC protocols apply |
| mCRPC-Liver metastasis |
Docetaxel;
ARPI (limited efficacy);
177Lu-PSMA-617 |
AFFIRM [165], PREVAIL [166], VISION [168] |
Poor prognosis;
AR loss, NE phenotype, high genomic instability; low PSMA expression limits radioligand use |
Novel ligands (e.g., FAP-PET) [169], PARPi [170,171], immunotherapy + TGF-β inhibition [172–174] |
| NEPC subtype |
Platinum chemo for NEPC (cisplatin + etoposide or carboplatin + docetaxel); SLC16LD6.5 for DLL3-expressing NEPC (Preclinical); AURKA inhibitor (Preclinical) |
NEPC trials [15,20,131,148,175,176] |
Insufficient objective response rate |
NEPC specific target selection; improve drug safety and efficacy |
DNPC subtype
(under research) |
FGFR inhibitor;
Gremlin1 antibody |
DNPC research [18,177,178] |
Lacks AR and NE markers; subset with FGFR activation |
Molecularly targeted trials; FGFR pathway inhibition |
| Novel therapy (under research/preclinical) |
AR targeted; non-AR targeted; DDR targeted; Immunotherapy |
RIPTAC [163], PROTAC [164]; EZH2 [179,180]; PROPEL [181], MAGNITUDE [182], TALAPRO-2 [183]; T-cell engagers, vaccines, CAR-T [184] |
Insufficient efficacy or toxicity concerns |
More specific biomarkers targets and safety |
This table summarizes current treatment strategies and challenges in mPCa patients with nonbone metastasis (lung, liver), focusing on both mCSPC and mCRPC stages. Treatment selection depends on site-specific biology, AR signaling status, subtype features (e.g., NEPC, DNPC), and novel therapy
References
| 1. |
Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. Ca-cancer J Clin. 2024;74:12-49.
[Google Scholar]
[CrossRef]
|
| 2. |
Bray F, Laversanne M, Sung HYA, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J Clin. 2024;74(3):229-263.
[Google Scholar]
[CrossRef]
|
| 3. |
Han BF, Zheng RS, Zeng HM, Wang SM, Sun KX, Chen R, et al. Cancer incidence and mortality in China, 2022. J Natl Cancer Inst. 2024;4(1):47-53.
[Google Scholar]
[CrossRef]
|
| 4. |
Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers. 2021;7:9.
[Google Scholar]
[CrossRef]
|
| 5. |
Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6(3):173.
[Google Scholar]
[CrossRef]
|
| 6. |
Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018;15(5):271-286.
[Google Scholar]
[CrossRef]
|
| 7. |
Liu JZ, Dong L, Zhu YJ, Dong BJ, Sha JJ, Zhu HH, et al. Prostate cancer treatment-China's perspective. Cancer Lett. 2022;550:215927.
[Google Scholar]
[CrossRef]
|
| 8. |
Akhtar M, Haider A, Rashid S, Al-Nabet A. Paget's "Seed and Soil" Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv Anat Pathol. 2019;26(1):69-74.
[Google Scholar]
[CrossRef]
|
| 9. |
Liu Y, Cao XT. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell. 2016;30(5):668-681.
[Google Scholar]
[CrossRef]
|
| 10. |
Gandaglia G, Abdollah F, Schiffmann J, Trudeau V, Shariat SF, Kim SP, et al. Distribution of Metastatic Sites in Patients With Prostate Cancer: A Population-Based Analysis. Prostate. 2014;74(2):210-216.
[Google Scholar]
[CrossRef]
|
| 11. |
Ma B, Wells A, Wei L, Zheng JN. Prostate cancer liver metastasis: Dormancy and resistance to therapy. Semin Cancer Biol. 2021;71:2-9.
[Google Scholar]
[CrossRef]
|
| 12. |
Li SL, Kang Y, Zeng Y. Targeting tumor and bone microenvironment: Novel therapeutic opportunities for castration-resistant prostate cancer patients with bone metastasis. Biochim Biophys Acta Rev Cancer. 2024;1879(1):189033.
[Google Scholar]
[CrossRef]
|
| 13. |
Logothetis C, Morris MJ, Den R, Coleman RE. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev. 2018;37(1):189-196.
[Google Scholar]
[CrossRef]
|
| 14. |
Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest. 2019;129(10):4492-4505.
[Google Scholar]
[CrossRef]
|
| 15. |
Wang Y, Wang Y, Ci XP, Choi SYC, Crea F, Lin D, et al. Molecular events in neuroendocrine prostate cancer development. Nat Rev Urol. 2021;18(10):581-596.
[Google Scholar]
[CrossRef]
|
| 16. |
Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298-305.
[Google Scholar]
[CrossRef]
|
| 17. |
Su WJ, Han HH, Wang Y, Zhang BY, Zhou B, Cheng YM, et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell. 2019;36(2):139-155.e10.
[Google Scholar]
[CrossRef]
|
| 18. |
Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32(4):474-489.e6.
[Google Scholar]
[CrossRef]
|
| 19. |
Lundberg A, Zhang M, Aggarwal R, Li HL, Zhang L, Foye A, et al. The Genomic and Epigenomic Landscape of Double-Negative Metastatic Prostate Cancer. Cancer Res. 2023;83(16):2763-2774.
[Google Scholar]
[CrossRef]
|
| 20. |
Conteduca V, Oromendia C, Eng KW, Bareja R, Sigouros M, Molina A, et al. Clinical features of neuroendocrine prostate cancer. Eur J Cancer. 2019;121:7-18.
[Google Scholar]
[CrossRef]
|
| 21. |
Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011-1025.
[Google Scholar]
[CrossRef]
|
| 22. |
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161(5):1215-1228.
[Google Scholar]
[CrossRef]
|
| 23. |
Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated Evolution of Prostate Cancer Genomes. Cell. 2013;153(3):666-677.
[Google Scholar]
[CrossRef]
|
| 24. |
Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao YH, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111(30):11139-11144.
[Google Scholar]
[CrossRef]
|
| 25. |
Akhoundova D, Francica P, Rottenberg S, Rubin MA. DNA Damage Response and Mismatch Repair Gene Defects in Advanced and Metastatic Prostate Cancer. Adv Anat Pathol. 2024;31(2):61-69.
[Google Scholar]
[CrossRef]
|
| 26. |
Pritchard CC, Offit K, Nelson PS. DNA-Repair Gene Mutations in Metastatic Prostate Cancer Reply. N Engl J Med. 2016;375(18):1804-1805.
[Google Scholar]
[CrossRef]
|
| 27. |
Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, et al. Oncogenic Genomic Alterations, Clinical Phenotypes, and Outcomes in Metastatic Castration-Sensitive for Prostate Cancer. Clin Cancer Res. 2020;26(13):3230-3238.
[Google Scholar]
[CrossRef]
|
| 28. |
Mateo J, Seed G, Bertan C, Rescigno P, Dolling D, Figueiredo I, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest. 2020;130(4):1743-1751.
[Google Scholar]
[CrossRef]
|
| 29. |
Rodriguez-Calero A, Gallon J, Akhoundova D, Maletti S, Ferguson A, Cyrta J, et al. Alterations in homologous recombination repair genes in prostate cancer brain metastases. Nat Commun. 2022;13:2400.
[Google Scholar]
[CrossRef]
|
| 30. |
Gallon J, Rodriguez-Calero A, Benjak A, Akhoundova D, Maletti S, Amstutz U, et al. DNA Methylation Landscapes of Prostate Cancer Brain Metastasis Are Shaped by Early Driver Genetic Alterations. Cancer Res. 2023;83(8):1203-1213.
[Google Scholar]
[CrossRef]
|
| 31. |
de Oliveira-Barros EG, Branco LC, Da Costa NM, Nicolau-Neto P, Palmero C, Pontes B, et al. GLIPR1 and SPARC expression profile reveals a signature associated with prostate Cancer Brain metastasis. Mol Cell Endocrinol. 2021;528:111230.
[Google Scholar]
[CrossRef]
|
| 32. |
Alshalalfa M, Seldon C, Franco I, Vince R, Carmona R, Punnen S, et al. Clinicogenomic characterization of prostate cancer liver metastases. Prostate Cancer Prostatic Dis. 2022;25(2):366-369.
[Google Scholar]
[CrossRef]
|
| 33. |
Feng E, Rydzewski NR, Zhang M, Lundberg A, Bootsma M, Helzer KT, et al. Intrinsic Molecular Subtypes of Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2022;28(24):5396-5404.
[Google Scholar]
[CrossRef]
|
| 34. |
Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428-11436.
[Google Scholar]
[CrossRef]
|
| 35. |
Ku SY, Rosario S, Wang YQ, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78-83.
[Google Scholar]
[CrossRef]
|
| 36. |
Zhou ZX, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66(16):7889-7898.
[Google Scholar]
[CrossRef]
|
| 37. |
Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004;64(17):6018-6025.
[Google Scholar]
[CrossRef]
|
| 38. |
Ludwig ML, Moline D, Horrmann A, Boytim E, Larson G, Arafa AT, et al. Integrated multi-omics assessment of lineage plasticity in a prostate cancer patient with brain and dural metastases. NPJ Precis Oncol. 2024;8:215.
[Google Scholar]
[CrossRef]
|
| 39. |
Ni XD, Wei Y, Li XM, Pan J, Fang BW, Zhang TW, et al. From biology to the clinic - exploring liver metastasis in prostate cancer. Nat Rev Urol. 2024;21(10):593-614.
[Google Scholar]
[CrossRef]
|
| 40. |
Shrestha R, Chesner LN, Zhang M, Zhou SL, Foye A, Lundberg A, et al. An Atlas of Accessible Chromatin in Advanced Prostate Cancer Reveals the Epigenetic Evolution during Tumor Progression. Cancer Res. 2024;84(18):3086-3100.
[Google Scholar]
[CrossRef]
|
| 41. |
Kim WK, Buckley AJ, Lee DH, Hiroto A, Nenninger CH, Olson AW, et al. Androgen deprivation induces double-null prostate cancer via aberrant nuclear export and ribosomal biogenesis through HGF and Wnt activation. Nat Commun. 2024;15:1231.
[Google Scholar]
[CrossRef]
|
| 42. |
Limberger T, Schlederer M, Trachtová K, Alonso IGD, Yang JY, Högler S, et al. KMT2C methyltransferase domain regulated INK4A expression suppresses prostate cancer metastasis. Mol Cancer. 2022;21:89.
[Google Scholar]
[CrossRef]
|
| 43. |
Cai HQ, Zhang B, Ahrenfeldt J, Joseph JV, Riedel M, Gao ZL, et al. CRISPR/Cas9 model of prostate cancer identifies Kmt2c deficiency as a metastatic driver by Odam/Cabs1 gene cluster expression. Nat Commun. 2024;15:2088.
[Google Scholar]
[CrossRef]
|
| 44. |
Guo J, Li N, Liu Q, Hao Z, Zhu G, Wang X, et al. KMT2C deficiency drives transdifferentiation of double-negative prostate cancer and confer resistance to AR-targeted therapy. Cancer Cell. 2025;43(7):1261-1278.e10.
[Google Scholar]
[CrossRef]
|
| 45. |
Cackowski FC, Kumar-Sinha C, Mehra R, Wu Y-M, Robinson DR, Alumkal JJ, et al. Double-Negative Prostate Cancer Masquerading as a Squamous Cancer of Unknown Primary: A Clinicopathologic and Genomic Sequencing-Based Case Study. JCO Precis Oncol. 2020;4:PO.20.00309.
[Google Scholar]
[CrossRef]
|
| 46. |
Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646-674.
[Google Scholar]
[CrossRef]
|
| 47. |
Ahmad F, Cherukuri MK, Choyke PL. Metabolic reprogramming in prostate cancer. Br J Cancer. 2021;125(9):1185-1196.
[Google Scholar]
[CrossRef]
|
| 48. |
Bader DA, McGuire SE. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat Rev Urol. 2020;17(4):214-231.
[Google Scholar]
[CrossRef]
|
| 49. |
Du A, Wang Z, Huang TD, Xue S, Jiang C, Qiu GT, et al. Fatty acids in cancer: Metabolic functions and potential treatment. Medcomm-Oncol. 2023;2(1):e25.
[Google Scholar]
[CrossRef]
|
| 50. |
Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2011;223(2):283-294.
[Google Scholar]
[CrossRef]
|
| 51. |
Pulliam TL, Awad D, Han JJ, Murray MM, Ackroyd JJ, Goli P, et al. Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer. Cells. 2022;11(12):1890.
[Google Scholar]
[CrossRef]
|
| 52. |
Chen YH, Chen H, Lin TT, Zhu JM, Chen JY, Dong RN, et al. ARPC1A correlates with poor prognosis in prostate cancer and is up-regulated by glutamine metabolism to promote tumor cell migration, invasion and cytoskeletal changes. Cell biosci. 2023;13:38.
[Google Scholar]
[CrossRef]
|
| 53. |
He YM, Ji ZZ, Gong YM, Fan LC, Xu PH, Chen XY, et al. Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell Rep. 2023;42(2):112033.
[Google Scholar]
[CrossRef]
|
| 54. |
Jing N, Zhang K, Chen XY, Liu KY, Wang JM, Xiao LL, et al. ADORA2A-driven proline synthesis triggers epigenetic reprogramming in neuroendocrine prostate and lung cancers. J Clin Invest. 2023;133(24):e168670.
[Google Scholar]
[CrossRef]
|
| 55. |
Samarzija I. Site-Specific and Common Prostate Cancer Metastasis Genes as Suggested by Meta-Analysis of Gene Expression Data. Life-Basel. 2021;11(7):636.
[Google Scholar]
[CrossRef]
|
| 56. |
Halabi S, Kelly WK, Ma H, Zhou HJ, Solomon NC, Fizazi K, et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J Clin Oncol. 2016;34(14):1652-1659.
[Google Scholar]
[CrossRef]
|
| 57. |
Tsilimigras DI, Brodt P, Clavien PA, Muschel RJ, D'Angelica MI, Endo I, et al. Liver metastases. Nat Rev Dis Primers. 2021;7:27.
[Google Scholar]
[CrossRef]
|
| 58. |
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after "Carcinoid": Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
[Google Scholar]
[CrossRef]
|
| 59. |
Liu K, Zhang Y, Du G, Chen X, Xiao L, Jiang L, et al. 5-HT orchestrates histone serotonylation and citrullination to drive neutrophil extracellular traps and liver metastasis. J Clin Invest. 2025;135(8):e183544.
[Google Scholar]
[CrossRef]
|
| 60. |
Deng YQ, Chen QC, Guo CY, Chen JH, Li X, Li ZY, et al. Comprehensive single-cell atlas of colorectal neuroendocrine tumors with liver metastases: unraveling tumor microenvironment heterogeneity between primary lesions and metastases. Mol Cancer. 2025;24:28.
[Google Scholar]
[CrossRef]
|
| 61. |
Chen XY, Bao W, Liu KY, Jing N, Du GY, Jiang LY, et al. O-GalNAc Glycosylation Activates MBL-Mediated Complement and Coagulation Cascades to Drive Organotropic Metastasis. Adv Sci. 2025;e04809.
[Google Scholar]
[CrossRef]
|
| 62. |
Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol. 2018;36:247-277.
[Google Scholar]
[CrossRef]
|
| 63. |
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306-321.
[Google Scholar]
[CrossRef]
|
| 64. |
Li J, Liu XG, Ge RL, Yin YP, Liu YD, Lu WP, et al. The ligation between ERMAP, galectin-9 and dectin-2 promotes Kupffer cell phagocytosis and antitumor immunity. Nat Immunol. 2023;24:1813-1824.
[Google Scholar]
[CrossRef]
|
| 65. |
Thomas SK, Wattenberg MM, Choi-Bose S, Uhlik M, Harrison B, Coho H, et al. Kupffer cells prevent pancreatic ductal adenocarcinoma metastasis to the liver in mice. Nat Commun. 2023;14:6330.
[Google Scholar]
[CrossRef]
|
| 66. |
Terabe M, Berzofsky JA. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front Immunol. 2018;9:1838.
[Google Scholar]
[CrossRef]
|
| 67. |
Jameson G, Harmon C, Santiago RM, Houlihan DD, Gallagher TK, Lynch L, et al. Human Hepatic CD56bright NK Cells Display a Tissue-Resident Transcriptional Profile and Enhanced Ability to Kill Allogenic CD8+ T Cells. Front Immunol. 2022;13:921212.
[Google Scholar]
[CrossRef]
|
| 68. |
Correia AL. Locally sourced: site-specific immune barriers to metastasis. Nat Rev Immunol. 2023;23(8):522-538.
[Google Scholar]
[CrossRef]
|
| 69. |
Crosby CM, Kronenberg M. Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol. 2018;18(9):559-574.
[Google Scholar]
[CrossRef]
|
| 70. |
Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931.
[Google Scholar]
[CrossRef]
|
| 71. |
Giannou AD, Kempski J, Shiri AM, Luecke J, Zhang T, Zhao L, et al. Tissue resident iNKT17 cells facilitate cancer cell extravasation in liver metastasis via interleukin-22. Immunity. 2023;56(1):125-142.e12.
[Google Scholar]
[CrossRef]
|
| 72. |
Peng H, Jiang XJ, Chen YL, Sojka DK, Wei HM, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. 2013;123(4):1444-1456.
[Google Scholar]
[CrossRef]
|
| 73. |
Lopes N, Galluso J, Escalière B, Carpentier S, Kerdiles YM, Vivier E. Tissue-specific transcriptional profiles and heterogeneity of natural killer cells and group 1 innate lymphoid cells. Cell Rep Med. 2022;3(11):100812.
[Google Scholar]
[CrossRef]
|
| 74. |
López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell. 2017;32(2):135-154.
[Google Scholar]
[CrossRef]
|
| 75. |
Zhang S, Liu WJ, Hu BW, Wang P, Lv X, Chen SF, et al. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front Immunol. 2020;11:1242.
[Google Scholar]
[CrossRef]
|
| 76. |
Correia AL, Guimaraes JC, Auf Der Maur P, De Silva D, Trefny MP, Okamoto R, et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature. 2021;594(7864):566-571.
[Google Scholar]
[CrossRef]
|
| 77. |
Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45(1):60-71.
[Google Scholar]
[CrossRef]
|
| 78. |
Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian ZG, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435-452.
[Google Scholar]
[CrossRef]
|
| 79. |
Shen K, Zheng S-S, Park O, Wang H, Sun Z, Gao B. Activation of innate immunity (NK/IFN-γ) in rat allogeneic liver transplantation: contribution to liver injury and suppression of hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G1070-G1077.
[Google Scholar]
[CrossRef]
|
| 80. |
Ducimetière L, Lucchiari G, Litscher G, Nater M, Heeb L, Nuñez NG, et al. Conventional NK cells and tissue-resident ILC1s join forces to control liver metastasis. Proc Natl Acad Sci U S A. 2021;118(27):e2026271118.
[Google Scholar]
[CrossRef]
|
| 81. |
Russo E, D'Aquino C, Di Censo C, Laffranchi M, Tomaipitinca L, Licursi V, et al. Cxcr3 promotes protection from colorectal cancer liver metastasis by driving NK cell infiltration and plasticity. J Clin Invest. 2025;135(11):e184036.
[Google Scholar]
[CrossRef]
|
| 82. |
Yan JX, Zhang C, Xu YL, Huang ZH, Ye QY, Qian XJ, et al. GPR34 is a metabolic immune checkpoint for ILC1-mediated antitumor immunity. Nat Immunol. 2024;25(11):2057-2067.
[Google Scholar]
[CrossRef]
|
| 83. |
Kondo T, Okabayashi K, Hasegawa H, Tsuruta M, Shigeta K, Kitagawa Y. The impact of hepatic fibrosis on the incidence of liver metastasis from colorectal cancer. Br J Cancer. 2016;115(1):34-39.
[Google Scholar]
[CrossRef]
|
| 84. |
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang HY, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816-826.
[Google Scholar]
[CrossRef]
|
| 85. |
Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211(8):1503-1523.
[Google Scholar]
[CrossRef]
|
| 86. |
James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med. 2017;377(4):338-351.
[Google Scholar]
[CrossRef]
|
| 87. |
Colomba E, Marret G, Baciarello G, Lavaud P, Massard C, Loriot Y, et al. Liver tests increase on abiraterone acetate in men with metastatic prostate cancer: Natural history, management and outcome. Eur J Cancer. 2020;129:117-122.
[Google Scholar]
[CrossRef]
|
| 88. |
Yun GY, Kim SH, Kim SW, Joo JS, Kim JS, Lee ES, et al. Atypical onset of bicalutamide-induced liver injury. World J Gastroenterol. 2016;22(15):4062-4065.
[Google Scholar]
[CrossRef]
|
| 89. |
Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med. 2017;377(4):352-360.
[Google Scholar]
[CrossRef]
|
| 90. |
Wang S, Friedman SL. Hepatic fibrosis: A convergent response to liver injury that is reversible. J Hepatol. 2020;73(1):210-211.
[Google Scholar]
[CrossRef]
|
| 91. |
Buxton AK, Abbasova S, Bevan CL, Leach DA. Liver Microenvironment Response to Prostate Cancer Metastasis and Hormonal Therapy. Cancers. 2022;14(24):6189.
[Google Scholar]
[CrossRef]
|
| 92. |
Kanda T, Yokosuka O. The androgen receptor as an emerging target in hepatocellular carcinoma. J Hepatocell Carcinoma. 2015;2:91-99.
[Google Scholar]
[CrossRef]
|
| 93. |
Hintz HM, Gallant JP, Vander Griend DJ, Coleman IM, Nelson PS, LeBeau AM. Imaging Fibroblast Activation Protein Alpha Improves Diagnosis of Metastatic Prostate Cancer with Positron Emission Tomography. Clin Cancer Res. 2020;26(18):4882-4891.
[Google Scholar]
[CrossRef]
|
| 94. |
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol. 2010;10(8):554-567.
[Google Scholar]
[CrossRef]
|
| 95. |
Drake CG. Visceral Metastases and Prostate Cancer Treatment: 'Die Hard,' 'Tough Neighborhoods,' or 'Evil Humors'? Oncology-NY. 2014;28(11):974-980.
[Google Scholar]
|
| 96. |
Yates CC, Shepard CR, Stolz DB, Wells A. Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer. 2007;96(8):1246-1252.
[Google Scholar]
[CrossRef]
|
| 97. |
Wells A, Yates C, Shepard CR. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis. 2008;25(6):621-628.
[Google Scholar]
[CrossRef]
|
| 98. |
Ma B, Wheeler SE, Clark AM, Whaley DL, Yang M, Wells A. Liver protects metastatic prostate cancer from induced death by activating E-cadherin signaling. Hepatology. 2016;64(5):1725-1742.
[Google Scholar]
[CrossRef]
|
| 99. |
Greenberg NM, Demayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, et al. PROSTATE-CANCER IN A TRANSGENIC MOUSE. Proc Natl Acad Sci U S A. 1995;92(8):3439-3443.
[Google Scholar]
[CrossRef]
|
| 100. |
Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED. The TRAMP mouse as a model for prostate cancer. Current protocols in immunology. 2001;45:20.5.1-20.5.23.
[Google Scholar]
[CrossRef]
|
| 101. |
Wang SY, Gao J, Lei QY, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4(3):209-221.
[Google Scholar]
[CrossRef]
|
| 102. |
Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, et al. Prostate Epithelial Pten/TP53 Loss Leads to Transformation of Multipotential Progenitors and Epithelial to Mesenchymal Transition. American J Pathol. 2011;179(1):422-435.
[Google Scholar]
[CrossRef]
|
| 103. |
Chen ZB, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725-730.
[Google Scholar]
[CrossRef]
|
| 104. |
Cho HJ, Herzka T, Zheng W, Qi J, Wilkinson JE, Bradner JE, et al. RapidCaP, a Novel GEM Model for Metastatic Prostate Cancer Analysis and Therapy, Reveals Myc as a Driver of Pten-Mutant Metastasis. Cancer Discov. 2014;4(3):318-333.
[Google Scholar]
[CrossRef]
|
| 105. |
Lau WM, Weber KL, Doucet M, Chou YT, Brady K, Kowalski J, et al. Identification of prospective factors promoting osteotropism in breast cancer: a potential role for CITED2. Int J Cancer. 2010;126(4):876-884.
[Google Scholar]
[CrossRef]
|
| 106. |
Qi XQ, Schepers E, Avella D, Kimchi ET, Kaifi JT, Staveley-O'Carroll KF, et al. An Oncogenic Hepatocyte-Induced Orthotopic Mouse Model of Hepatocellular Cancer Arising in the Setting of Hepatic Inflammation and Fibrosis. Jove-J Vis Exp. 2019;151:10.3791/59368.
[Google Scholar]
[CrossRef]
|
| 107. |
Stuparu AD, Meyer CAL, Evans-Axelsson SL, Lückerath K, Wei LH, Kim W, et al. Targeted alpha therapy in a systemic mouse model of prostate cancer - a feasibility study. Theranostics. 2020;10(6):2612-2620.
[Google Scholar]
[CrossRef]
|
| 108. |
Simons BW, Dalrymple S, Rosen M, Zheng L, Brennen WN. A hemi-spleen injection model of liver metastasis for prostate cancer. Prostate. 2020;80(14):1263-1269.
[Google Scholar]
[CrossRef]
|
| 109. |
Liu KY, Jing N, Wang D, Xu PH, Wang JM, Chen XY, et al. A novel mouse model for liver metastasis of prostate cancer reveals dynamic tumour-immune cell communication. Cell Prolif. 2021;54(7):e13056.
[Google Scholar]
[CrossRef]
|
| 110. |
Sailer V, von Amsberg G, Duensing S, Kirfel J, Lieb V, Metzger E, et al. Experimental in vitro, ex vivo and in vivo models in prostate cancer research. Nat Rev Urol. 2023;20(3):158-178.
[Google Scholar]
[CrossRef]
|
| 111. |
Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell. 2014;159(1):176-187.
[Google Scholar]
[CrossRef]
|
| 112. |
Cheaito K, Bahmad H, Hadadeh O, Msheik H, Monzer A, Ballout F, et al. Establishment and characterization of prostate organoids from treatment-naive patients with prostate cancer. Oncol Lett. 2022;23(1):6.
[Google Scholar]
[CrossRef]
|
| 113. |
Wang YW, Xue H, Cutz JC, Bayani J, Mawji NR, Chen WG, et al. An orthotopic metastatic prostate cancer model in SCID mice via grafting of a transplantable human prostate tumor line. Lab Invest. 2005;85(11):1392-1404.
[Google Scholar]
[CrossRef]
|
| 114. |
Önner H, Özer H, Çelik AV, Yilmaz F, Gedik GK. Isolated Liver Metastasis Detected by 68Ga-PSMA PET/CT in Newly Diagnosed Prostate Cancer. Clin Nucl Med. 2023;48(3):259-260.
[Google Scholar]
[CrossRef]
|
| 115. |
De Man K, Van Laeken N, Schelfhout V, Fendler WP, Lambert B, Kersemans K, et al. 18F-PSMA-11 Versus 68Ga-PSMA-11 Positron Emission Tomography/ Computed Tomography for Staging and Biochemical Recurrence of Prostate Cancer: A Prospective Double-blind Randomised Cross-over Trial. Eur Urol. 2022;82(5):501-509.
[Google Scholar]
[CrossRef]
|
| 116. |
Seniaray N, Verma R, Belho E, Malik D, Mahajan H. Diffuse Pulmonary Metastases From Prostate Cancer on 68Ga PSMA PET/CT. Clin Nucl Med. 2019;44(11):898-900.
[Google Scholar]
[CrossRef]
|
| 117. |
Soydal C, Ozkan E, Yerlikaya H, Utkan G, Kucuk ON. Widespread Metastatic Prostate Carcinoma Shown by 68Ga-PSMA PET/CT. Clin Nucl Med. 2016;41(6):E294-E295.
[Google Scholar]
[CrossRef]
|
| 118. |
Chan M, Hsiao E, Turner J. LCerebellar Metastases From Prostate Cancer on 68Ga-PSMA PET/CT. Clin Nucl Med. 2017;42(3):193-194.
[Google Scholar]
[CrossRef]
|
| 119. |
Seifert R, Emmett L, Rowe SP, Herrmann K, Hadaschik B, Calais J, et al. Second Version of the Prostate Cancer Molecular Imaging Standardized Evaluation Framework Including Response Evaluation for Clinical Trials (PROMISE V2). Eur Urol. 2023;83(5):405-412.
[Google Scholar]
[CrossRef]
|
| 120. |
Damjanovic J, Janssen JC, Prasad V, Diederichs G, Walter T, Brenner W, et al. 68Ga-PSMA-PET/CT for the evaluation of liver metastases in patients with prostate cancer. Canc Imaging. 2019;19:37.
[Google Scholar]
[CrossRef]
|
| 121. |
Watt F, Martorana A, Brookes DE, Ho T, Kingsley E, O'Keefe DS, et al. A tissue-specific enhancer of the prostate-specific membrane antigen gene, FOLH1. Genomics. 2001;73(3):243-254.
[Google Scholar]
[CrossRef]
|
| 122. |
Noss KR, Wolfe SA, Grimes SR. Upregulation of prostate specific membrane antigen/folate hydrolase transcription by an enhancer. Gene. 2002;285(1-2):247-256.
[Google Scholar]
[CrossRef]
|
| 123. |
Jadvar H. Molecular imaging of prostate cancer with 18F-fluorodeoxyglucose PET. Nat Rev Urol. 2009;6(6):317-323.
[Google Scholar]
[CrossRef]
|
| 124. |
Spratt DE, Gavane S, Tarlinton L, Fareedy SB, Doran MG, Zelefsky MJ, et al. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate. 2014;74(11):1153-1159.
[Google Scholar]
[CrossRef]
|
| 125. |
Bakht MK, Lovnicki JM, Tubman J, Stringer KF, Chiaramonte J, Reynolds MR, et al. Differential Expression of Glucose Transporters and Hexokinases in Prostate Cancer with a Neuroendocrine Gene Signature: A Mechanistic Perspective for 18F-FDG Imaging of PSMA-Suppressed Tumors. J Nucl Med. 2020;61(6):904-910.
[Google Scholar]
[CrossRef]
|
| 126. |
Ahumada JV, Rueda SDG, Solís FSA, Cortés QP, Agredo LTP, Miguel JR, et al. Multitarget Molecular Imaging in Metastatic Castration Resistant Adenocarcinoma Prostate Cancer with Therapy Induced Neuroendocrine Differentiation. Diagnostics. 2022;12(6):1387.
[Google Scholar]
[CrossRef]
|
| 127. |
Korsen JA, Kalidindi TM, Khitrov S, Samuels ZV, Chakraborty G, Gutierrez JA, et al. Molecular Imaging of Neuroendocrine Prostate Cancer by Targeting Delta-Like Ligand 3. J Nucl Med. 2022;63(9):1401-1407.
[Google Scholar]
[CrossRef]
|
| 128. |
Tendler S, Dunphy MP, Agee M, O'Donoghue J, Aly RG, Choudhury NJ, et al. Imaging with [89Zr]Zr-DFO-SC16.56 anti-DLL3 antibody in patients with high-grade neuroendocrine tumours of the lung and prostate: a phase 1/2, first-in-human trial. Lancet Oncol. 2024;25(8):1015-1024.
[Google Scholar]
[CrossRef]
|
| 129. |
Taranda J, Mathew G, Watrud K, El-Amine N, Lee MTF, Elowsky C, et al. Combined whole-organ imaging at single-cell resolution and immunohistochemical analysis of prostate cancer and its liver and brain metastases. Cell Rep. 2021;37(7):110027.
[Google Scholar]
[CrossRef]
|
| 130. |
Berchuck JE, Viscuse PV, Beltran H, Aparicio A. Clinical considerations for the management of androgen indifferent prostate cancer. Prostate Cancer Prostatic Dis. 2021;24(3):623-637.
[Google Scholar]
[CrossRef]
|
| 131. |
Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang XM, Reyes A, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol. 2002;20(14):3072-3080.
[Google Scholar]
[CrossRef]
|
| 132. |
Aggarwal R, Huang JT, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol. 2018;36(24):2492-2503.
[Google Scholar]
[CrossRef]
|
| 133. |
Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol. 2021;18(5):297-312.
[Google Scholar]
[CrossRef]
|
| 134. |
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164(1-2):57-68.
[Google Scholar]
[CrossRef]
|
| 135. |
Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10(466):eaat4921.
[Google Scholar]
[CrossRef]
|
| 136. |
Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016;2(12):1598-1606.
[Google Scholar]
[CrossRef]
|
| 137. |
Volik S, Alcaide M, Morin RD, Collins C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol Cancer Res. 2016;14(10):898-908.
[Google Scholar]
[CrossRef]
|
| 138. |
Ulz P, Belic J, Graf R, Auer M, Lafer I, Fischereder K, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Commun. 2016;7:12008.
[Google Scholar]
[CrossRef]
|
| 139. |
Romanel A, Tandefelt DG, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7(312):312re10.
[Google Scholar]
[CrossRef]
|
| 140. |
Kumar A, Coleman I, Morrissey C, Zhang XT, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369-378.
[Google Scholar]
[CrossRef]
|
| 141. |
Hong MKH, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605.
[Google Scholar]
[CrossRef]
|
| 142. |
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353-357.
[Google Scholar]
[CrossRef]
|
| 143. |
Grasso CS, Wu YM, Robinson DR, Cao XH, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239-243.
[Google Scholar]
[CrossRef]
|
| 144. |
Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J Natl Cancer Inst. 2018;109(12):djx118.
[Google Scholar]
[CrossRef]
|
| 145. |
Schweizer MT, Sivakumar S, Tukachinsky H, Coleman I, De Sarkar N, Yu EY, et al. Concordance of DNA Repair Gene Mutations in Paired Primary Prostate Cancer Samples and Metastatic Tissue or Cell-Free DNA. JAMA Oncol. 2021;7(9):1378-1382.
[Google Scholar]
[CrossRef]
|
| 146. |
Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest. 2020;130(4):1653-1668.
[Google Scholar]
[CrossRef]
|
| 147. |
Franceschini GM, Quaini O, Mizuno K, Orlando F, Ciani Y, Ku SY, et al. Noninvasive Detection of Neuroendocrine Prostate Cancer through Targeted Cell-free DNA Methylation. Cancer Discov. 2024;14(3):424-445.
[Google Scholar]
[CrossRef]
|
| 148. |
Aparicio AM, Harzstark AL, Corn PG, Wen SJ, Araujo JC, Tu SM, et al. Platinum-Based Chemotherapy for Variant Castrate-Resistant Prostate Cancer. Clin Cancer Res. 2013;19(13):3621-3630.
[Google Scholar]
[CrossRef]
|
| 149. |
Brimo F, Epstein JI. Immunohistochemical pitfalls in prostate pathology. Hum Pathol. 2012;43(3):313-324.
[Google Scholar]
[CrossRef]
|
| 150. |
Yamada Y, Beltran H. The treatment landscape of metastatic prostate cancer. Cancer Lett. 2021;519:20-29.
[Google Scholar]
[CrossRef]
|
| 151. |
Posdzich P, Darr C, Hilser T, Wahl M, Herrmann K, Hadaschik B, et al. Metastatic Prostate Cancer-A Review of Current Treatment Options and Promising New Approaches. Cancers. 2023;15(2):461.
[Google Scholar]
[CrossRef]
|
| 152. |
Akaza H, Yamaguchi A, Matsuda T, Igawa M, Kumon H, Soeda A, et al. Superior anti-tumor efficacy of bicalutamide 80 mg in combination with a luteinizing hormone-releasing hormone (LHRH) agonist versus LHRH agonist monotherapy as first-line treatment for advanced prostate cancer: Interim results of a randomized study in Japanese patients. Japanese J Clin Oncol. 2004;34(1):20-28.
[Google Scholar]
[CrossRef]
|
| 153. |
Deegen P, Thomas O, Nolan-Stevaux O, Li S, Wahl J, Bogner P, et al. The PSMA-targeting Half-life Extended BiTE Therapy AMG 160 has Potent Antitumor Activity in Preclinical Models of Metastatic Castration-resistant Prostate Cancer. Clin Cancer Res. 2021;27(10):2928-2937.
[Google Scholar]
[CrossRef]
|
| 154. |
Corn PG, Heath EI, Zurita A, Ramesh N, Xiao LC, Sei E, et al. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: a randomised, open-label, phase 1-2 trial. Lancet Oncol. 2019;20(10):1432-1443.
[Google Scholar]
[CrossRef]
|
| 155. |
Yamada Y, Beltran H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr Oncol Rep. 2021;23(2):15.
[Google Scholar]
[CrossRef]
|
| 156. |
Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N Engl J Med. 2019;380(13):1235-1246.
[Google Scholar]
[CrossRef]
|
| 157. |
Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N Engl J Med. 2018;378(15):1408-1418.
[Google Scholar]
[CrossRef]
|
| 158. |
Lowrance WT, Breau RH, Chou R, Chapin BF, Crispino T, Dreicer R, et al. Advanced Prostate Cancer: AUA/ASTRO/SUO Guideline PART I. J Urol. 2021;205(1):14-21.
[Google Scholar]
[CrossRef]
|
| 159. |
Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med. 2015;373(8):737-746.
[Google Scholar]
[CrossRef]
|
| 160. |
James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387(10024):1163-1177.
[Google Scholar]
[CrossRef]
|
| 161. |
Fizazi K, Foulon S, Carles J, Roubaud G, McDermott R, Fléchon A, et al. Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castration-sensitive prostate cancer (PEACE-1): a multicentre, open-label, randomised, phase 3 study with a 2 x 2 factorial design. Lancet. 2022;399(10336):1695-1707.
[Google Scholar]
[CrossRef]
|
| 162. |
Hussain M, Tombal B, Saad F, Fizazi K, Sternberg CN, Crawford ED, et al. Darolutamide Plus Androgen-Deprivation Therapy and Docetaxel in Metastatic Hormone-Sensitive Prostate Cancer by Disease Volume and Risk Subgroups in the Phase III ARASENS Trial. J Clin Oncol. 2023;41(20):3595-3607.
[Google Scholar]
[CrossRef]
|
| 163. |
Raina K, Eastman KJ, Yu X, Forbes CD, Jones KM, Mousseau JJ, et al. An oral androgen receptor RIPTAC for prostate cancer. J Clin Oncol. 2023;41:184.
[Google Scholar]
[CrossRef]
|
| 164. |
Zhang YL, Ming AN, Wang JY, Chen WM, Fang ZQ. PROTACs targeting androgen receptor signaling: Potential therapeutic agents for castration-resistant prostate cancer. Pharmacol Res. 2024;205:107234.
[Google Scholar]
[CrossRef]
|
| 165. |
Alumkal JJ, Chowdhury S, Loriot Y, Sternberg CN, de Bono JS, Tombal B, et al. Effect of Visceral Disease Site on Outcomes in Patients With Metastatic Castration-resistant Prostate Cancer Treated With Enzalutamide in the PREVAIL Trial. Clin Genitourin Cancer. 2017;15(5):610-617.e3.
[Google Scholar]
[CrossRef]
|
| 166. |
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N Engl J Med. 2014;371(5):424-433.
[Google Scholar]
[CrossRef]
|
| 167. |
Loriot Y, Fizazi K, de Bono JS, Forer D, Hirmand M, Scher HI. Enzalutamide in Castration-Resistant Prostate Cancer Patients With Visceral Disease in the Liver and/or Lung: Outcomes From the Randomized Controlled Phase 3 AFFIRM Trial. Cancer. 2017;123(2):253-262.
[Google Scholar]
[CrossRef]
|
| 168. |
Sartor O, de Bono J, Chi KN, Fizazi K, Herrmann K, Rahbar K, et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2021;385(12):1091-1103.
[Google Scholar]
[CrossRef]
|
| 169. |
Kessel K, Seifert R, Weckesser M, Boegemann M, Huss S, Kratochwil C, et al. Prostate-specific membrane antigen and fibroblast activation protein distribution in prostate cancer: preliminary data on immunohistochemistry and PET imaging. Ann Nucl Med. 2022;36(3):293-301.
[Google Scholar]
[CrossRef]
|
| 170. |
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;382(22):2091-2102.
[Google Scholar]
[CrossRef]
|
| 171. |
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J Clin Oncol. 2020;38(32):3763-3772.
[Google Scholar]
[CrossRef]
|
| 172. |
Yu JL, Green MD, Li SS, Sun YL, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27(1):152-164.
[Google Scholar]
[CrossRef]
|
| 173. |
Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T Cell Surveillance of Oncogene-Induced Prostate Cancer Is Impeded by T Cell-Derived TGF-β1 Cytokine. Immunity. 2011;35(1):123-134.
[Google Scholar]
[CrossRef]
|
| 174. |
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538-543.
[Google Scholar]
[CrossRef]
|
| 175. |
Puca L, Gavyert K, Sailer V, Conteduca V, Dardenne E, Sigouros M, et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci Transl Med. 2019;11(484):eaav0891.
[Google Scholar]
[CrossRef]
|
| 176. |
Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res. 2019;25(1):43-51.
[Google Scholar]
[CrossRef]
|
| 177. |
Cheng CP, Wang JM, Xu PH, Zhang K, Xin ZX, Zhao HF, et al. Gremlin1 is a therapeutically targetable FGFR1 ligand that regulates lineage plasticity and castration resistance in prostate cancer. Nat Cancer. 2022;3(5):565-580.
[Google Scholar]
[CrossRef]
|
| 178. |
Sena LA, Brennen WN, Isaacs JT. There are gremlins in prostate cancer. Nat Cancer. 2022;3(5):530-531.
[Google Scholar]
[CrossRef]
|
| 179. |
Schweizer MT, Penkov K, Choudhury AD, Calvo E, Frank RC, Liu L, et al. Phase 1 trial of mevrometostat (PF-06821497), a potent and selective inhibitor of enhancer of zeste homolog 2 (EZH2), in castration-resistant prostate cancer (CRPC). J Clin Oncol. 2024;42:5061.
[Google Scholar]
[CrossRef]
|
| 180. |
Calvo M, Penkov K, Spira AI, Candilejo IM, Shore ND, Zhang T, et al. A multi-center, open-label, randomized dose expansion study of PF-06821497, a potent and selective inhibitor of enhancer of zeste homolog 2 (EZH2), in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2023;41:TPS282.
[Google Scholar]
[CrossRef]
|
| 181. |
Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial (vol 24, pg 1094, 2023). Lancet Oncol. 2024;25(5):e180-e180.
[Google Scholar]
[CrossRef]
|
| 182. |
Chi KN, Sandhu S, Smith MR, Attard G, Saad M, Olmos D, et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: second interim analysis of the randomized phase III MAGNITUDE trial. Ann Oncol. 2023;34(9):772-782.
[Google Scholar]
[CrossRef]
|
| 183. |
Fizazi K, Azad AA, Matsubara N, Carles J, Fay AP, De Giorgi U, et al. First-line talazoparib with enzalutamide in HRR-deficient metastatic castration-resistant prostate cancer: the phase 3 TALAPRO-2 trial (vol 30, pg 257, 2024). Nat Med. 2024;30(5):1505.
[Google Scholar]
[CrossRef]
|
| 184. |
Cimadamore A, Boixareu C, Sharp A, Beltran H, de Bono JS. Novel Therapeutic Strategies for Metastatic Prostate Cancer Care. Eur Urol. 2025;19:S0302-838(25)00357-4.
[Google Scholar]
[CrossRef]
|
| 185. |
Pond GR, Sonpavde G, de Wit R, Eisenberger MA, Tannock IF, Armstrong AJ. The Prognostic Importance of Metastatic Site in Men with Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2014;65(1):3-6.
[Google Scholar]
[CrossRef]
|
| 186. |
Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the TAX 327 study. J Clin Oncol. 2008;26(2):242-245.
[Google Scholar]
[CrossRef]
|
| 187. |
Pouessel D, Gallet B, Bibeau F, Avancès C, Iborra F, Sénesse P, et al. Liver metastases in prostate carcinoma: clinical characteristics and outcome. Bju Int. 2007;99(4):807-811.
[Google Scholar]
[CrossRef]
|
| 188. |
Ku SY, Gleave ME, Beltran H. Towards precision oncology in advanced prostate cancer. Nat Rev Urol. 2019;16(11):645-654.
[Google Scholar]
[CrossRef]
|
| 189. |
Alabi BR, Liu SQ, Stoyanova T. Current and emerging therapies for neuroendocrine prostate cancer. Pharmacol Ther. 2022;238:108255.
[Google Scholar]
[CrossRef]
|
| 190. |
Gerstberger S, Jiang QW, Ganesh K. Metastasis. Cell. 2023;186(8):1564-1579.
[Google Scholar]
[CrossRef]
|
| 191. |
de Visser KE, Joyce JA. The evolving tumor microenvironment From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41(3):374-403.
[Google Scholar]
[CrossRef]
|
| 192. |
Tang L, Peng SC, Zhuang XW, He Y, Song YX, Nie H, et al. Tumor Metastasis: Mechanistic Insights and Therapeutic Intervention. Medcomm-Oncol. 2025;4(1):e70012.
[Google Scholar]
[CrossRef]
|
| 193. |
Coleman RE, Croucher PI, Padhani AR, Clézardin P, Chow E, Fallon M, et al. Bone metastases. Nat Rev Dis Primers. 2020;6:83.
[Google Scholar]
[CrossRef]
|
| 194. |
Wang WC, Yang X, Dai JL, Lu Y, Zhang J, Keller ET. Prostate cancer promotes a vicious cycle of bone metastasis progression through inducing osteocytes to secrete GDF15 that stimulates prostate cancer growth and invasion. Oncogene. 2019;38(23):4540-4559.
[Google Scholar]
[CrossRef]
|
| 195. |
Zhang WJ, Bado IL, Hu JY, Wan YW, Wu L, Wang H, et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell. 2021;184(9):2471-2486.e20.
[Google Scholar]
[CrossRef]
|
| 196. |
Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer. 2020;1(7):672-680.
[Google Scholar]
[CrossRef]
|
| 197. |
Ikawa K, Terashima Y, Sasaki K, Tashiro S. Genetic detection of liver micrometastases that are undetectable histologically. J Surg Res. 2002;106(1):124-130.
[Google Scholar]
[CrossRef]
|
| 198. |
Noltenius C, Noltenius H. DORMANT TUMOR-CELLS IN LIVER AND BRAIN - AN AUTOPSY STUDY ON METASTASIZING TUMORS. Pathol Res Pract. 1985;179(4-5):504-511.
[Google Scholar]
[CrossRef]
|
| 199. |
He D, Wu Q, Tian P, Liu Y, Jia Z, Li Z, et al. Chemotherapy awakens dormant cancer cells in lung by inducing neutrophil extracellular traps. Cancer Cell. 2025;S1535-6108(25):00257-0.
[Google Scholar]
[CrossRef]
|
| 200. |
Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM. Pulmonary Alveolar Macrophages Contribute to the Premetastatic Niche by Suppressing Antitumor T Cell Responses in the Lungs. J Immunol. 2015;194(11):5529-5538.
[Google Scholar]
[CrossRef]
|
| 201. |
Liu YF, Gu Y, Han YM, Zhang Q, Jiang ZP, Zhang X, et al. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell. 2016;30(2):243-256.
[Google Scholar]
[CrossRef]
|
| 202. |
March B, Faulkner S, Jobling P, Steigler A, Blatt A, Denham J, et al. Tumour innervation and neurosignalling in prostate cancer. Nat Rev Urol. 2020;17(2):119-130.
[Google Scholar]
[CrossRef]
|
| 203. |
Mancusi R, Monje M. The neuroscience of cancer. Nature. 2023;618(7965):467-479.
[Google Scholar]
[CrossRef]
|
| 204. |
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136-153.
[Google Scholar]
[CrossRef]
|

 ,
Penghui Xu
,
Penghui Xu