Abstract
Progression through the mammalian cell cycle is a highly regulated process to maintain tissue homeostasis. The key regulators of cell cycle transitions are cyclin-dependent kinase (CDK)/Cyclin complexes that phosphorylate substrates such as the RB tumor suppressor to facilitate cellular division. The regulation of G1/S is of particular significance in cancer and is affected by numerous tumor suppressors and oncogenes. Historically, the cell cycle was viewed as a rigidly regulated process, but recent evidence has revealed significant flexibility and differential CDK/Cyclin dependencies across tumor types. These heterogeneous features of cell cycle control have implications for the etiology of different tumor types as well as the response to multiple therapeutic modalities. Most notably, adaptive responses in cell cycle regulatory circuits can contribute to acquired resistance in a variety of contexts, underscoring the importance for tumor biology and disease treatment.
Keywords
Cell cycle, cancer, plasticity, cyclin, CDK, CDK inhibitor, palbociclib
1. Overview of G1/S Control
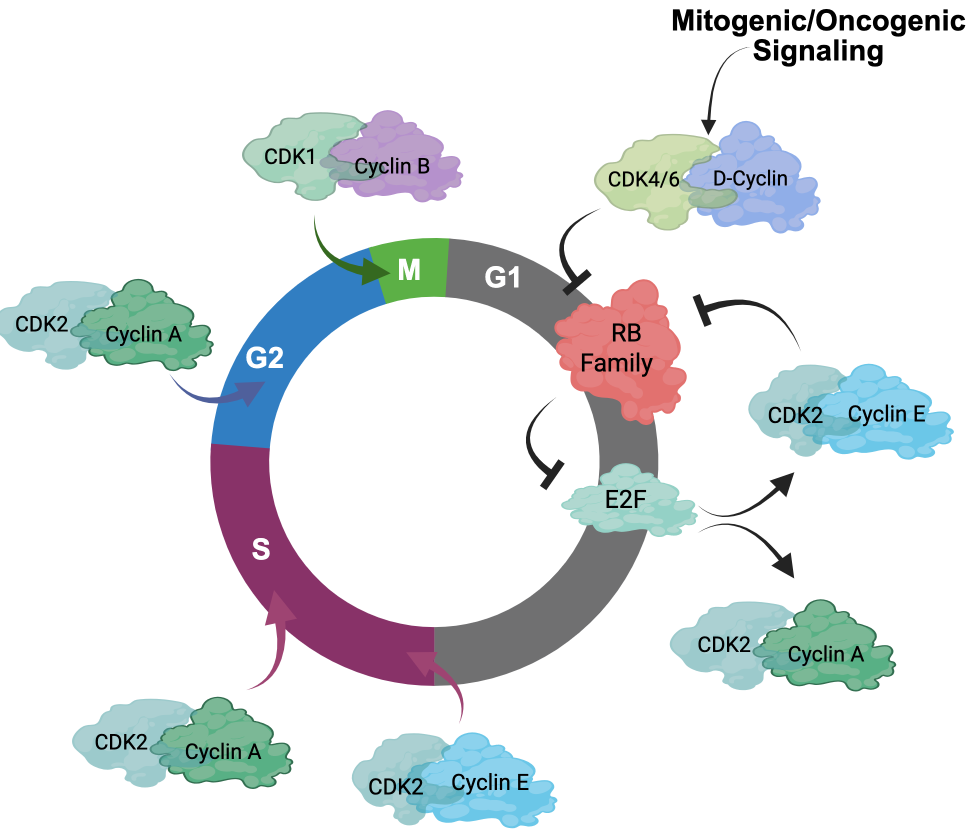
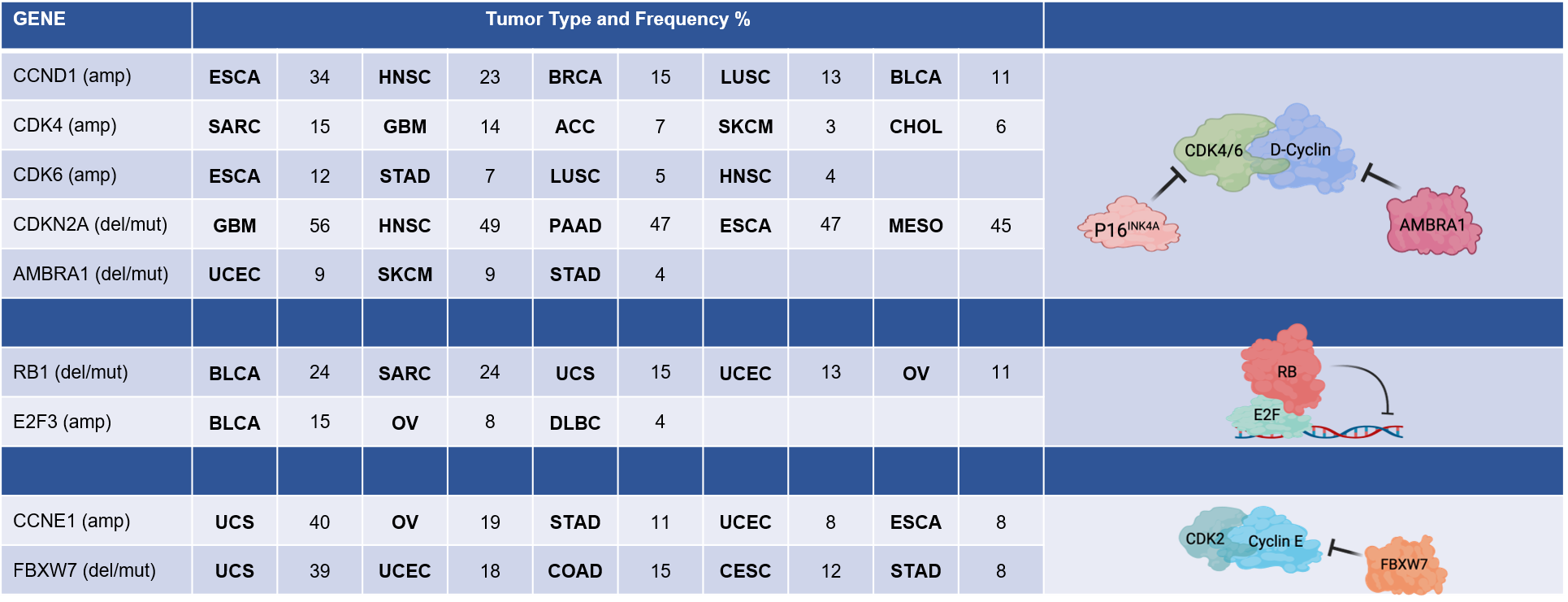
The key regulatory step controlled by oncogenic networks is traversal of the G1/S phase transition of the cell cycle [1]. Oncogenes such as KRAS, ERBB2, and EGFR highjack conventional mitogenic networks to promote cell cycle progression. Mitogens are believed to interface with the cell cycle through cyclin-dependent kinase (CDK) 4/6 and D-type cyclin complexes (Figure 1) [1–4]. Growth factors induce the expression of D-type cyclins (Cyclin D1, D2, or D3) in a variety of tissues and promote the stabilization and activation of CDK4/6 kinases through multiple mechanisms [3]. The importance of CDK4/6 in tumor development is evidenced by the frequent amplification or aberration of CDK4, CDK6, and CCND1 encoding Cyclin D1 in many cancers (Figure 2) [5–7]. Conversely, negative regulators of Cyclin D1 or CDK4/6 are tumor suppressors. For example, CDKN2A, which encodes the tumor suppressor p16INK4A, is a negative regulator of CDK4/6 and is frequently lost in many cancers (Figure 2) [8–10]. The AMBRA1 tumor suppressor mediates the degradation of Cyclin D1 [11,12] (Figure 2). Thus, it has been presumed that just as KRAS mutations bypass dependence on mitogenic signals, alterations in the cell cycle bypass upstream processes that enable unregulated proliferation.
A key substrate of CDK4 and CDK6 activity is the retinoblastoma tumor suppressor (RB) [4,14,15]. RB was the first tumor suppressor identified and, along with its related pocket protein family members p107 and p130, conventionally serves to coordinate gene expression programs controlled by the E2F transcription factor (Figure 1 and 2) [16–18]. The E2F transcriptional program encodes many “pan-essential” genes that promote DNA replication (e.g., MCM7, CDC6, CDT1) and mitotic progression (e.g., PLK1, AURKA, CCNB1) [17,19–22]. In its active, hypophosphorylated state, RB binds E2F and serves as a potent transcriptional co-repressor [23,24]. Phosphorylation of RB, as well as p107 and p130, by CDKs releases RB from E2F and alleviates this transcriptional repression, enabling progression through G1/S [17,25]. A number of E2F genes are amplified in cancers including E2F3 and E2F1 [26,27]. Many targeted therapies act to prevent RB phosphorylation and, thus, repress this central transcriptional circuit. As would be expected, this RB/E2F transcriptional circuit is commonly deregulated in cancer and is generally associated with cancers that have poor prognosis [28,29].
Another driver of the G1/S transition is Cyclin E, encoded by CCNE1 [30–32]. Like CCND1, CCNE1 amplification occurs in several cancer types (Figure 2). Cyclin E interacts with CDK2 and is believed to play an important role in driving G1/S progression as well as coordinating initiation of DNA replication and transit through G2 phase of the cell cycle [32,33] (Figure 1). The regulation of Cyclin E is complex and involves transcriptional and post-translational modifications that modulate protein accumulation. Some of these processes are related to oncogenesis, as the tumor suppressor FBXW7 modulates Cyclin E stability as one of its targets [34]. Cyclin E is considered an E2F-regulated gene and has been proposed to function at least in part downstream of RB [35,36]. However, Cyclin E and CDK2 activity is also mitogen regulated, albeit the mechanisms are not as clearly understood as the mitogen regulation of CDK4/6. Yet, one notable mechanism is the expression of p21Cip1 and p27Kip1, which are endogenous CDK2 inhibitors, that are induced by a variety of signaling events and serve to constrain CDK2 activity [37–39].
2. Cell Cycle Definitions
With thousands of review articles published showing a common cell cycle regulatory structure, terms like plasticity and heterogeneity may seem out of place. The term plasticity is defined as the quality of being easily shaped or molded. In a biological sense, this would include alterations in a particular process or mechanism either occurring stochastically or under particular stressors. In one sense, plasticity and evolution are related, with a definition of evolution being the gradual development of something, especially from a simple to a more complex form. Perhaps the time scale is different but conceptually both share similarities in changes occurring from an initial primordial state. From a cell cycle perspective, such plasticity or evolution could be from using CDK4 to initiate the cell cycle to using CDK2, or perhaps the evolution to an RB-deficient state under selective pressure. Data from mouse models with CDK or Cyclin deletions indicate that such transitions can occur under the pressure of organismal survival. Heterogeneity in contrast reflects an end state; the quality or state of being diverse in character or content. For example, many solid cancers have heterogeneity at diagnosis in terms of predominant CDK/Cyclin or RB status associated with a given cancer. As indicated above, this heterogeneity can be related to the genetics of the tumor, such as amplifications and epigenetic features. These definitions are important as plasticity and evolution ultimately promote heterogeneity and these processes are continual during cancer etiology, tumor progression, and acquired resistance to therapy. In addition to states that promote tumor cell proliferation, as is the focus here, there are clearly other adaptations such as tumor dormancy [40], polyploid giant cells [41], whole-genome doubling [42], and other cell cycle processes that may ultimately lead to increased fitness as a result of alterations in cell cycle control.
3. Heterogeneous Nature of Cancer Cell Cycles
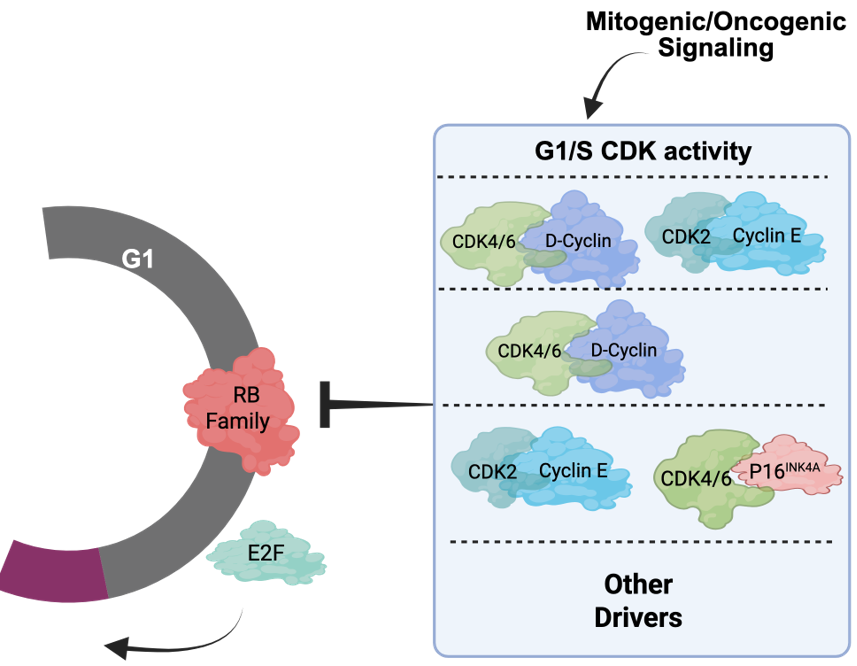
While the cell cycle is often drawn, by us and others, as a circle with different CDK and cyclin activities promoting various transitions (e.g., Figure 1) [43, 44], this summation belies the intrinsic complexity of cell cycle and redundancies in most facets of G1/S control [45,46]. Based on biochemical activities, it would be expected that many cell cycle drivers such as CDK4 or CDK2 would be pan-essential. However, mice can develop normally, for the most part, in the absence of CDK2 and develop with tissue-selective deficits upon CDK4 or CDK6 deletion [47,48]. While the combination of CDK4 and CDK6 deletion is lethal in mice, it has a limited impact on the development of multiple cell lineages, with these cells successfully progressing through the cycle [49]. In fact, it is possible to ablate the “interphase” CDKs (CDK4, CDK6, and CDK2) to yield a cell cycle that is driven by CDK1 in mice [50]. In the context of tumor models such plasticity is also apparent where Cyclin D3 can compensate for Cyclin D1 deletion in ERBB2 mammary tumorigenesis [51]. Thus, there is significant redundancy relative to individual CDK genes and an inherent ability to adapt and use “non-canonical” CDK/Cyclin complexes to facilitate G1/S transition.
Large-scale CRISPR screens in cancer cells have further expanded our understanding of key CDK and cyclin gene dependencies [52,53]. These data suggest that the importance of a given CDK or cyclin is associated with a complex interaction between cancer genetics, epigenetic states, and features that likely pertain to tumor lineage. For example, dependence on CDK4 is associated with amplification of CCND1 or expression of the Cyclin D1 protein [52,53]. In contrast, dependence on CDK2 is associated with high expression of Cyclin E and the expression of p16INK4A, limiting the ability of these cells to use CDK4/6 for cell cycle progression [54–57]. In this context, cell lineage is also an important feature related to cell cycle control. For example, multiple hematological cancers preferentially utilize CDK6 for proliferation, which is consistent with CDK6 use in normal hematopoietic progenitor proliferation as well [49,58,59]. Further, advancement of single-cell analyses has provided the opportunity to track these dependencies at higher resolution over time to help define the alterations of cell cycle drivers in a given tumor [60–65]. For example, single cell analyses can illustrate rapid adaptation to CDK4/6 inhibition which can occur through CDK2/Cyclin D1 complexes [66] and yield enhanced sensitivity to CDK2 inhibition [62].
Taking these data into consideration, there is perhaps no singular canonical cancer cell cycle, but rather different tumors have a preferred cell cycle for the G1/S transition (Figure 3). This concept of varying cell cycle states allows individual tumors to have distinct cell cycles that are shaped by multiple features of that specific tumor. By interrogating many tumors, it is possible to start to infer common paths that may be relevant for tumor etiology or therapeutic responses. For example, many ovarian tumors express high levels of Cyclin E and p16INK4A with relatively low levels of Cyclin D1 [54–57]. This configuration would predict more dependence on CDK2. In contrast, most hormone receptor-positive, human epidermal growth factor receptor 2-negative (HR+/HER2-) breast cancer presentations have high expression of D-type cyclins (i.e., Cyclin D1), with low Cyclin E and p16INK4A that could be associated with sensitivity to CDK4/6 inhibitors that are FDA approved for that tumor type [46,52,67]. While these heterogeneous cell cycle states can pre-exist as indicated above, they can adapt to various stresses including therapies and thus are not static.
4. Cell Cycle Deregulation in Cancer
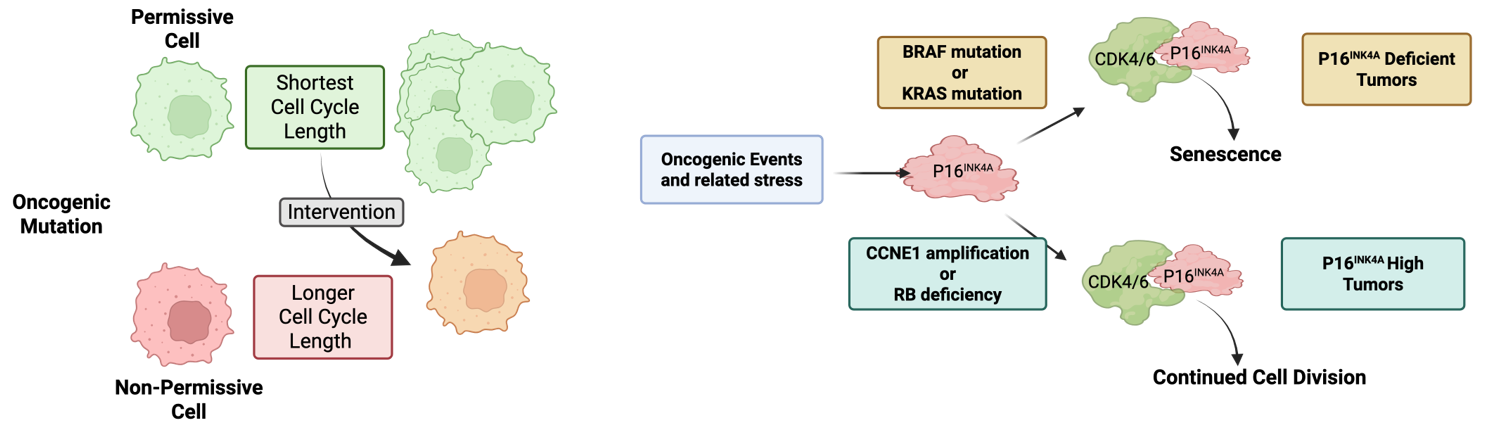
As noted above, several key mediators of G1/S control are oncogenes and tumor suppressors. Since many of these genes are in fact multi-functional, there remains controversy related to whether cell cycle control and proliferation or other processes are key in driving tumor development. For example, while Cyclin E overexpression can advance cell cycle transitions, Cyclin E can exert replication stress and features of chromosome instability that could contribute to cancer etiology [32,68]. Similarly, while RB deficiency deregulates cell cycle, RB loss has broad effects related to lineage states and epigenetic programs beyond cell cycle [18,44,69]. Such complexities are perhaps part of the reason why most cell cycle regulators have a limited spectrum of relevance in disease etiology. For example, if RB is a global mediator of cell cycle control, then why is germline RB loss associated primarily with pediatric retinoblastoma, with secondary malignancies (e.g., soft-tissue sarcoma and osteosarcoma) emerging sporadically and much later in life [70]? Similarly, germline CDKN2A deficiency (while in the same pathway as RB) is associated with familial atypical multiple mole melanoma syndrome that yields increased risk for specifically melanoma and pancreatic cancer [71]. A new study interrogating the cell of origin for RB1 deficiency showed that a key determinant of tissue tropism is the ability of select cells to deregulate cell cycle transit [72], defining the cells of origin as having the shortest cell cycle length (Figure 4A). By a variety of genetic permutations/interventions, it is possible to restrict tumor development by lengthening cell cycle transit [72]. This study employed multiple oncogenic mutations beyond RB1 loss and indicates that the tumor spectrum is highly related to those cells that are most susceptible to a given perturbation changing cell cycle transit times to fuel tumor development.
Outside of direct permutations to the cell cycle machinery, veritably all cancers are characterized by classic cancer hallmarks such as mitogen-independent growth and escape from proliferative limitations, among others [73]. In the context of many oncogenic drivers, there are specific mechanisms through which they promote deregulation of cell cycle (e.g., upregulation of Cyclin D1 and depletion of p27Kip1) [74, 75]. Oncogenes in the RAS-BRAF-MAPK pathway can potently drive proliferation but also have the capacity to drive oncogene-induced senescence [76,77] (Figure 4B). This particular response to oncogenic signaling is caused at least in part by the upregulation of p16INK4A, which inhibits CDK4/6 to limit cell cycle progression [77,78]. This phenomenon is likely very effective in limiting tumor development, as benign nevi that are driven by mutant BRAF are arrested in G1 by p16INK4A and can remain in that non-malignant state [79]. Presumably, this is a relatively general mechanism to suppress oncogenesis and likely explains why genetic or epigenetic loss of the CDKN2A locus is a particularly common event in tumors driven through this KRAS-BRAF-MAPK axis (Figure 4B). Interestingly, this induction of CDKN2A also occurs with oncogenes that intrinsically bypass the requirement for CDK4/6 [80,81]. For example, tumors driven by RB loss or CCNE1 amplification almost invariably overexpress p16INK4A [56,82,83]. This is likely an epigenetic vestige of the oncogenic stress mediated by loss of RB or Cyclin E gain; however, in this context, cell division can continue with high p16INK4A (Figure 4B).
5. Therapy Resistance with Targeted Therapy
Most therapies that were developed to target oncogenic or mitogenic signaling (e.g., EGFR, KRAS, and MEK inhibitors; endocrine therapy) have their predominant effect by arresting cells in a G1 or G0-like state [43,75,84–86]. This is because, in effect, these agents are cutting off the supply of mitogens. In a variety of contexts, this is associated with subsequent death of tumor cells as the oncogenic signal is required for viability as well as proliferation. However, particularly in advanced solid tumors, residual arrested tumor cells survive. The response of these cells is dependent upon the disease paradigm and, thus, is highly varied, including responses such as therapy-induced quiescent/dormant/senescent cells, disease-tolerant persisters, minimal residual disease cells, etc. [87–90]. Irrespective of the terminology used to describe them, these cells are functionally arrested with a 2N DNA content, with classical features of cell cycle inhibition (e.g., dephosphorylation of RB) but serve as a reservoir from which acquired resistance can emerge. Invariably the re-emergence of disease or progression of stable disease involves the ability of sub-clones of the original tumor population to re-enter the cell cycle [87].
Cell cycle reactivation is highly dependent on the tumor type and therapy used. With many targeted therapies, the predominant mechanism driving acquired resistance is at the level of the target and bypass signaling [91]. For example, ESR1 and AR mutations and aberrations are selected during treatment of HR+/HER2- breast and prostate cancer with endocrine therapy [92–94]. Similarly, mutations of EGFR and RAS are relatively common means to bypass drug effects [95–97], spurring the development of multiple “next-generation” inhibitors [98]. Alternatively, signaling pathways can frequently restore mitogenic signaling to bypass selective inhibitors. While not as common, deregulation of the cell cycle through amplification of CDK/Cyclins or loss of RB1 have been observed following the development of resistance to targeted therapies. For example, a spectrum of non-small cell lung cancer and pancreatic ductal adenocarcinoma tumors harbor CDK or Cyclin amplifications associated with resistance to EGFR and KRAS inhibitors [99,100]. Conversely, RB deficiency is associated with resistance to endocrine therapy and targeted therapies in several paradigms. Specifically in the context of prostate cancer and non-small cell lung cancer, RB1 loss is associated with a lineage transition toward a neuroendocrine phenotype that not only deregulates the cell cycle but also diminishes the importance of EGFR or AR in the biology of these tumors [44,101–103]. This does not appear to be the case in other models (e.g., triple-negative breast cancer or bladder cancer), where RB deficiency is not linked to a neuroendocrine phenotype. Since so many mechanisms of resistance act upstream of cell cycle, there has been significant energy into directly inhibiting CDK activity.
6. Pharmacological CDK Inhibitors
Based on the mechanisms through which resistance to targeted therapies emerges, therapeutically targeting downstream nodes may be particularly effective. Selective CDK4/6 inhibitors were first reported over 20 years ago [104,105]. These agents exhibited efficacy in mantle cell lymphoma [106], which is driven by Cyclin D1. However, the major success of these drugs since has been in the context of HR+/HER2- breast cancer in combination with endocrine therapy [107–109]. Multiple clinical trials demonstrated substantial improvement in progression-free survival [110–112], and now CDK4/6 inhibitors are widely deployed in the treatment of breast cancer. The clinical success of these drugs in HR+/HER2- breast cancer has driven significant clinical testing. A review of clinicaltrials.gov using the search terms “palbociclib,” “ribociclib,” and “abemaciclib” returned 800 results that span most disease sites. Several recent reviews have tabulated these clinical trials with differing combination therapies [113–116]. Despite this vast amount of clinical research, the only FDA approvals for CDK4/6 inhibitors are in HR+/HER2- breast cancer. Why this is the case remains only partially understood.
While single agent CDK4/6 inhibitors at relatively high doses (e.g., >1 μM) can have a profound effect on many cell types, this dose is not consistent with clinical drug exposure levels. Even in preclinical models, many cells can adapt to treatment with CDK4/6 inhibitor monotherapy, including HR+/HER2- breast cancer [66,117,118]. The underlying adaptations often involve parallel CDK2 activity driving the cell cycle and bypassing the necessity of CDK4/6 activity [117,119]. This response can occur rapidly and limits the utility of CDK4/6 inhibitors. Genetic screens and analyses of acquired resistance have further illustrated that many events can mediate resistance to CDK4/6 inhibitors, typically acting by uncoupling RB activation from drug treatment [11,46,117,119–121].
In principle, the challenges of overcoming single agent resistance can be achieved through the use of combination therapies, and many are highly effective in preclinical models [120,122]. Indeed, most of the ongoing clinical trials employ some form of combination therapy. Where studied, the mechanism of cooperation is typically through impacting adaptive resistance mechanisms and inducing what is considered a “deeper” cell cycle arrest that sometimes associates with a senescent-like phenotype [123–125]. The process is modulated at least in part by the TP53 tumor suppressor [126], which indirectly controls CDK2 activity and further supports the combination with CDK2 inhibitors as discussed in more detail below. Whether ongoing clinical studies will yield more FDA approvals for CDK4/6 inhibitors remains unknown, but it is likely that CDK4/6 inhibition will be broadly considered in combination strategies moving forward.
Due to the success and challenges of CDK4/6 inhibitors, new strategies are emerging to more effectively target the cell cycle machinery therapeutically. First, one approach is to limit the toxicity of the inhibitors in the clinic, thereby enabling higher on-target activity and emulating what is observed with higher drug doses in preclinical models. The dose-limiting toxicity for palbociclib and ribociclib is neutropenia, which is believed to be due to the key role of CDK6 in features of hematopoiesis [59,127]. Therefore, drugs have been developed that are more selective for CDK4 [128]. The agent atirmociclib can be dosed at a higher level in preclinical studies, enabling better disease control [128]. Ongoing clinical trials are evaluating the ability of this CDK4-selective inhibitor to treat patients with HR+/HER2- breast cancer including those who progressed on the standard of care CDK4/6 inhibitors. Second, a number of CDK2 inhibitors have been developed. These agents have a very interesting mechanism of action and serve as a reminder of the importance of cancer genetics when considering targeting drivers of G1/S. CDK2 inhibitors are particularly active in tumors driven by Cyclin E, but are even more effective when both Cyclin E and p16INK4A, the endogenous CDK4/6 inhibitor, are highly expressed [54–57]. This molecular landscape creates a bottleneck for G1/S progression that is controlled by CDK2. Thus, ongoing clinical trials are testing CDK2 inhibitors specifically in CCNE1-amplified cancers [13]. Outside of tumors with this distinct cell cycle regulatory landscape, CDK2 inhibitors generally induce a G2 phase arrest [55]. In most models studied to date, there has been substantial synergy when using CDK2 and CDK4/6 inhibitors jointly, suggesting that there are ways to pharmacologically induce molecular bottlenecks similar to what is observed in CCNE1-amplified tumors [54–57,129]. While much of this work has involved catalytic inhibitors, CDK2-targeting PROTACs and other mechanisms to inhibit CDK activity are emerging and are currently being tested in early phase clinical trials, which have been recently reviewed [13]. Overall, the study of CDK inhibitors for cancer therapy have reinforced the plasticity and heterogeneity of cancer cell cycles.
7. Conclusions
While the convenient textbook depiction of cell cycle is likely here for perpetuity, heterogeneity and plasticity of cell cycle in tissues and tumors illustrates the importance of considering malleable cell cycle regulatory landscapes. This has significance relevant to the etiology of select tumors driven by permutations in the cell cycle, as well as adaptations that can occur in the context of resistance to targeted therapies.
Declarations
Ethics Statement
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Funding
This study was supported by funding to ESK and AKW from the NCI (CA247362 and CA247362-S1) and from the Roswell Park Alliance Foundation. This work was also supported by RPCCC and the NCI (P30CA016056).
Competing interests
ESK has sponsored research funded by Blueprint Medicine and Bristol Myers Squibb and is a member of the Cancer Cell Cycle-LLC consulting enterprise. AKW has sponsored research funded by Blueprint Medicine and Bristol Myers Squibb.
Acknowledgement
The authors thank their long-standing colleagues in the field for stimulating conversations through the years and regret any omissions.
Abbreviations
- CDK:
- Cyclin-dependent kinase
- HER2:
- Human epidermal growth factor receptor 2
- HR:
- Hormone receptor
References
| 1. |
Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73-92.
[Google Scholar]
|
| 2. |
Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65(4):701-713.
[Google Scholar]
[CrossRef]
|
| 3. |
Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20(5):187-190.
[Google Scholar]
[CrossRef]
|
| 4. |
Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14(3):2077-2086.
[Google Scholar]
[CrossRef]
|
| 5. |
Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A. 1994;91(2):709-713.
[Google Scholar]
[CrossRef]
|
| 6. |
Arnold A, Motokura T, Bloom T, Rosenberg C, Bale A, Kronenberg H, et al. PRAD1 (cyclin D1): a parathyroid neoplasia gene on 11q13. Henry Ford Hosp Med J. 1992;40(3–4):177-180.
[Google Scholar]
|
| 7. |
He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day RS 3rd, et al. CDK4 amplification is an alternative mechanism to p16 gene homozygous deletion in glioma cell lines. Cancer Res. 1994;54(22):5804-5807.
[Google Scholar]
|
| 8. |
Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11(2):210-212.
[Google Scholar]
[CrossRef]
|
| 9. |
Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704-707.
[Google Scholar]
[CrossRef]
|
| 10. |
Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27-32.
[Google Scholar]
[CrossRef]
|
| 11. |
Chaikovsky AC, Li C, Jeng EE, Loebell S, Lee MC, Murray CW, et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature. 2021;592(7856):794-798.
[Google Scholar]
[CrossRef]
|
| 12. |
Simoneschi D, Rona G, Zhou N, Jeong YT, Jiang S, Milletti G, et al. CRL4(AMBRA1) is a master regulator of D-type cyclins. Nature. 2021;592(7856):789-793.
[Google Scholar]
[CrossRef]
|
| 13. |
Knudsen ES, Witkiewicz AK, Sanidas I, Rubin SM. Targeting CDK2 for cancer therapy. Cell Rep. 2025;44(8):116140.
[Google Scholar]
[CrossRef]
|
| 14. |
Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672-1677.
[Google Scholar]
[CrossRef]
|
| 15. |
Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323-334.
[Google Scholar]
[CrossRef]
|
| 16. |
Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671-682.
[Google Scholar]
[CrossRef]
|
| 17. |
Fischer M, Schade AE, Branigan TB, Muller GA, DeCaprio JA. Coordinating gene expression during the cell cycle. Trends Biochem Sci. 2022;47(12):1009-1022.
[Google Scholar]
[CrossRef]
|
| 18. |
Goodrich DW. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene. 2006;25(38):5233-5243.
[Google Scholar]
[CrossRef]
|
| 19. |
Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19(6):658-662.
[Google Scholar]
[CrossRef]
|
| 20. |
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16(2):245-256.
[Google Scholar]
[CrossRef]
|
| 21. |
Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK. Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget. 2017;8(27):43678-43691.
[Google Scholar]
[CrossRef]
|
| 22. |
Markey MP, Angus SP, Strobeck MW, Williams SL, Gunawardena RW, Aronow BJ, et al. Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res. 2002;62(22):6587-6597.
[Google Scholar]
|
| 23. |
Kaelin WG Jr. Alterations in G1/S cell-cycle control contributing to carcinogenesis. Ann N Y Acad Sci. 1997;833:29-33.
[Google Scholar]
[CrossRef]
|
| 24. |
Sellers WR, Rodgers JW, Kaelin WG Jr. A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc Natl Acad Sci U S A. 1995;92(25):11544-11548.
[Google Scholar]
[CrossRef]
|
| 25. |
Cam H, Dynlacht BD. Emerging roles for E2F: Beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3(4):311-316.
[Google Scholar]
[CrossRef]
|
| 26. |
Oeggerli M, Tomovska S, Schraml P, Calvano-Forte D, Schafroth S, Simon R, et al. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene. 2004;23(33):5616-5623.
[Google Scholar]
[CrossRef]
|
| 27. |
Feber A, Clark J, Goodwin G, Dodson AR, Smith PH, Fletcher A, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004;23(8):1627-1630.
[Google Scholar]
[CrossRef]
|
| 28. |
Knudsen ES, Nambiar R, Rosario SR, Smiraglia DJ, Goodrich DW, Witkiewicz AK. Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun Biol. 2020;3(1):158.
[Google Scholar]
[CrossRef]
|
| 29. |
Iacovacci J, Brough R, Moughari FA, Alexander J, Kemp H, Tutt ANJ, et al. Proteogenomic discovery of RB1-defective phenocopy in cancer predicts disease outcome, response to treatment, and therapeutic targets. Sci Adv. 2025;11(13):eadq9495.
[Google Scholar]
[CrossRef]
|
| 30. |
Roberts JM, Koff A, Polyak K, Firpo E, Collins S, Ohtsubo M, et al. Cyclins, Cdks, and cyclin kinase inhibitors. Cold Spring Harb Symp Quant Biol. 1994;59:31-38.
[Google Scholar]
[CrossRef]
|
| 31. |
Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, et al. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66(6):1217-1228.
[Google Scholar]
[CrossRef]
|
| 32. |
Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776-2786.
[Google Scholar]
[CrossRef]
|
| 33. |
Chi Y, Carter JH, Swanger J, Mazin AV, Moritz RL, Clurman BE. A novel landscape of nuclear human CDK2 substrates revealed by in situ phosphorylation. Sci Adv. 2020;6(16):eaaz9899.
[Google Scholar]
[CrossRef]
|
| 34. |
Siu KT, Rosner MR, Minella AC. An integrated view of cyclin E function and regulation. Cell Cycle. 2012;11(1):57-64.
[Google Scholar]
[CrossRef]
|
| 35. |
DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15(8):4215-4224.
[Google Scholar]
[CrossRef]
|
| 36. |
Herrera RE, Sah VP, Williams BO, Makela TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16(5):2402-2407.
[Google Scholar]
[CrossRef]
|
| 37. |
Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78(1):59-66.
[Google Scholar]
[CrossRef]
|
| 38. |
Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8(1):9-22.
[Google Scholar]
[CrossRef]
|
| 39. |
Reed SI, Bailly E, Dulic V, Hengst L, Resnitzky D, Slingerland J. G1 control in mammalian cells. J Cell Sci Suppl. 1994;18:69-73.
[Google Scholar]
[CrossRef]
|
| 40. |
Agudo J, Aguirre-Ghiso JA, Bhatia M, Chodosh LA, Correia AL, Klein CA. Targeting cancer cell dormancy. Nat Rev Cancer. 2024;24(2):97-104.
[Google Scholar]
[CrossRef]
|
| 41. |
Liu J, Erenpreisa J, Sikora E. Polyploid giant cancer cells: An emerging new field of cancer biology. Sem Cancer Biol. 2022;81:1-4.
[Google Scholar]
[CrossRef]
|
| 42. |
Lambuta RA, Nanni L, Liu Y, Diaz-Miyar J, Iyer A, Tavernari D, et al. Whole-genome doubling drives oncogenic loss of chromatin segregation. Nature. 2023;615(7954):925-933.
[Google Scholar]
[CrossRef]
|
| 43. |
Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714-724.
[Google Scholar]
[CrossRef]
|
| 44. |
Knudsen ES, Pruitt SC, Hershberger PA, Witkiewicz AK, Goodrich DW. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer. 2019;5(5):308-324.
[Google Scholar]
[CrossRef]
|
| 45. |
Witkiewicz AK, Kumarasamy V, Sanidas I, Knudsen ES. Cancer cell cycle dystopia: heterogeneity, plasticity, and therapy. Trends Cancer. 2022;8(9):711-725.
[Google Scholar]
[CrossRef]
|
| 46. |
Knudsen ES, Witkiewicz AK, Rubin SM. Cancer takes many paths through G1/S. Trends Cell Biol. 2023;34(8):P636-645.
[Google Scholar]
[CrossRef]
|
| 47. |
Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13(20):1775-1785.
[Google Scholar]
[CrossRef]
|
| 48. |
Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22(1):44-52.
[Google Scholar]
[CrossRef]
|
| 49. |
Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493-504.
[Google Scholar]
[CrossRef]
|
| 50. |
Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448(7155):811-815.
[Google Scholar]
[CrossRef]
|
| 51. |
Zhang Q, Sakamoto K, Liu C, Triplett AA, Lin WC, Rui H, et al. Cyclin D3 compensates for the loss of cyclin D1 during ErbB2-induced mammary tumor initiation and progression. Cancer Res. 2011;71(24):7513-7524.
[Google Scholar]
[CrossRef]
|
| 52. |
Zhang Z, Golomb L, Meyerson M. Functional Genomic Analysis of CDK4 and CDK6 Gene Dependency across Human Cancer Cell Lines. Cancer Res. 2022;82(11):2171-2184.
[Google Scholar]
[CrossRef]
|
| 53. |
Knudsen ES, Kumarasamy V, Nambiar R, Pearson JD, Vail P, Rosenheck H, et al. CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep. 2022;38(9):110448.
[Google Scholar]
[CrossRef]
|
| 54. |
Sine C, Watts L, Fernandez B, Wang J, Knudsen E, Witkiewicz A, et al. p16 expression confers sensitivity to CDK2 inhibitors. bioRxiv. 2025.
[Google Scholar]
[CrossRef]
|
| 55. |
Kumarasamy V, Wang J, Roti M, Wan Y, Dommer AP, Rosenheck H, et al. Discrete vulnerability to pharmacological CDK2 inhibition is governed by heterogeneity of the cancer cell cycle. Nat Commun. 2025;16(1):1476.
[Google Scholar]
[CrossRef]
|
| 56. |
Dommer AP, Kumarasamy V, Wang J, O'Connor TN, Roti M, Mahan S, et al. Tumor Suppressors Condition Differential Responses to the Selective CDK2 Inhibitor BLU-222. Cancer Res. 2025;85(7):1310-1326.
[Google Scholar]
[CrossRef]
|
| 57. |
House NC, Brown VE, Chen M, Yuan L, Moore SL, Guo J, et al. Profiling the Activity of the Potent and Highly Selective CDK2 Inhibitor BLU-222 Reveals Determinants of Response in CCNE1-Aberrant Ovarian and Endometrial Tumors. Cancer Res. 2025;85(7):1297-1309.
[Google Scholar]
[CrossRef]
|
| 58. |
Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan AS, Prchal-Murphy M, Heller G, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125(1):90-101.
[Google Scholar]
[CrossRef]
|
| 59. |
Maurer B, Brandstoetter T, Kollmann S, Sexl V, Prchal-Murphy M. Inducible deletion of CDK4 and CDK6 - deciphering CDK4/6 inhibitor effects in the hematopoietic system. Haematologica. 2021;106(10):2624-2632.
[Google Scholar]
[CrossRef]
|
| 60. |
Stallaert W, Taylor SR, Kedziora KM, Taylor CD, Sobon HK, Young CL, et al. The molecular architecture of cell cycle arrest. Mol Syst Biol. 2022;18(9):e11087.
[Google Scholar]
[CrossRef]
|
| 61. |
Ranek JS, Stallaert W, Milner JJ, Redick M, Wolff SC, Beltran AS, et al. DELVE: feature selection for preserving biological trajectories in single-cell data. Nature communications. 2024;15(1):2765.
[Google Scholar]
[CrossRef]
|
| 62. |
Zikry TM, Wolff SC, Ranek JS, Davis HM, Naugle A, Luthra N, et al. Cell cycle plasticity underlies fractional resistance to palbociclib in ER+/HER2- breast tumor cells. Proc Natl Acad Sci U S A. 2024;121(7):e2309261121.
[Google Scholar]
[CrossRef]
|
| 63. |
Wen O, Wolff SC, Stallaert W, Li D, Purvis JE, Zikry TM. Spherical Manifolds Capture Drug-Induced Changes in Tumor Cell Cycle Behavior. Pac Symp Biocomput. 2025;30:473-487.
[Google Scholar]
[CrossRef]
|
| 64. |
Matson JP, Cook JG. Cell cycle proliferation decisions: the impact of single cell analyses. FEBS J. 2017;284(3):362-375.
[Google Scholar]
[CrossRef]
|
| 65. |
Riba A, Oravecz A, Durik M, Jimenez S, Alunni V, Cerciat M, et al. Cell cycle gene regulation dynamics revealed by RNA velocity and deep-learning. Nat Commun. 2022;13(1):2865.
[Google Scholar]
[CrossRef]
|
| 66. |
Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016.
[Google Scholar]
[CrossRef]
|
| 67. |
Palafox M, Monserrat L, Bellet M, Villacampa G, Gonzalez-Perez A, Oliveira M, et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER(+) breast cancer. Nat Commun. 2022;13(1):5258.
[Google Scholar]
[CrossRef]
|
| 68. |
Fagundes R, Teixeira LK. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front Cell Dev Biol. 2021;9:774845.
[Google Scholar]
[CrossRef]
|
| 69. |
Dick FA, Goodrich DW, Sage J, Dyson NJ. Non-canonical functions of the RB protein in cancer. Nat Rev Cancer. 2018;18(7):442-451.
[Google Scholar]
[CrossRef]
|
| 70. |
Schonfeld SJ, Kleinerman RA, Abramson DH, Seddon JM, Tucker MA, Morton LM. Long-term risk of subsequent cancer incidence among hereditary and nonhereditary retinoblastoma survivors. Br J Cancer. 2021;124(7):1312-1319.
[Google Scholar]
[CrossRef]
|
| 71. |
Danishevich A, Bilyalov A, Nikolaev S, Khalikov N, Isaeva D, Levina Y, et al. CDKN2A Gene Mutations: Implications for Hereditary Cancer Syndromes. Biomedicines. 2023;11(12):3343.
[Google Scholar]
[CrossRef]
|
| 72. |
Chen D, Lu S, Huang K, Pearson JD, Pacal M, Peidis P, et al. Cell cycle duration determines oncogenic transformation capacity. Nature. 2025;641(8065):1309-1318.
[Google Scholar]
[CrossRef]
|
| 73. |
Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46.
[Google Scholar]
[CrossRef]
|
| 74. |
Narita Y, Nagane M, Mishima K, Huang HJ, Furnari FB, Cavenee WK. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002;62(22):6764-6769.
[Google Scholar]
|
| 75. |
Isermann T, Sers C, Der CJ, Papke B. KRAS inhibitors: resistance drivers and combinatorial strategies. Trends Cancer. 2025;11(2):91-116.
[Google Scholar]
[CrossRef]
|
| 76. |
Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642.
[Google Scholar]
[CrossRef]
|
| 77. |
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593-602.
[Google Scholar]
[CrossRef]
|
| 78. |
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633-637.
[Google Scholar]
[CrossRef]
|
| 79. |
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720-724.
[Google Scholar]
[CrossRef]
|
| 80. |
Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10(15):2497-2503.
[Google Scholar]
[CrossRef]
|
| 81. |
Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, et al. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30(18):2087-2097.
[Google Scholar]
[CrossRef]
|
| 82. |
Bartkova J, Lukas J, Guldberg P, Alsner J, Kirkin AF, Zeuthen J, et al. The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56(23):5475-5483.
[Google Scholar]
|
| 83. |
Tort F, Bartkova J, Sehested M, Orntoft T, Lukas J, Bartek J. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 2006;66(21):10258-10263.
[Google Scholar]
[CrossRef]
|
| 84. |
Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631-643.
[Google Scholar]
[CrossRef]
|
| 85. |
Dutta PR, Maity A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007;254(2):165-177.
[Google Scholar]
[CrossRef]
|
| 86. |
Kumarasamy V, Wang J, Frangou C, Wan Y, Dynka A, Rosenheck H, et al. The Extracellular Niche and Tumor Microenvironment Enhance KRAS Inhibitor Efficacy in Pancreatic Cancer. Cancer Res. 2024;84(7):1115-1132.
[Google Scholar]
[CrossRef]
|
| 87. |
Soragni A, Knudsen ES, O'Connor TN, Tognon CE, Tyner JW, Gini B, et al. Acquired resistance in cancer: towards targeted therapeutic strategies. Nat Rev Cancer. 2025;25:613-633.
[Google Scholar]
[CrossRef]
|
| 88. |
Neel DS, Bivona TG. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis Oncol. 2017;1:3.
[Google Scholar]
[CrossRef]
|
| 89. |
Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst. 2021;113(10):1285-1298.
[Google Scholar]
[CrossRef]
|
| 90. |
Santos-de-Frutos K, Djouder N. When dormancy fuels tumour relapse. Commun Biol. 2021;4(1):747.
[Google Scholar]
[CrossRef]
|
| 91. |
Stubbs NM, Roady TJ, Schwermann MP, Eteshola EO, MacDonald WJ, Purcell C, et al. Acquired resistance to molecularly targeted therapies for cancer. Cancer Drug Resist. 2025;8:27.
[Google Scholar]
[CrossRef]
|
| 92. |
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell. 2018;34(3):427-438.e426.
[Google Scholar]
[CrossRef]
|
| 93. |
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446-1451.
[Google Scholar]
[CrossRef]
|
| 94. |
Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat Rev. 2017;57:16-27.
[Google Scholar]
[CrossRef]
|
| 95. |
Riedl JM, Fece de la Cruz F, Lin JJ, Parseghian C, Kim JE, Matsubara H, et al. Genomic landscape of clinically acquired resistance alterations in patients treated with KRAS(G12C) inhibitors. Ann Oncol. 2025;36(6):682-692.
[Google Scholar]
[CrossRef]
|
| 96. |
Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. 2021;384(25):2382-2393.
[Google Scholar]
[CrossRef]
|
| 97. |
Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, et al. The T790M "gatekeeper" mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7(4):874-879.
[Google Scholar]
[CrossRef]
|
| 98. |
Holderfield M, Lee BJ, Jiang J, Tomlinson A, Seamon KJ, Mira A, et al. Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy. Nature. 2024;629(8013):919-926.
[Google Scholar]
[CrossRef]
|
| 99. |
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725-737.
[Google Scholar]
[CrossRef]
|
| 100. |
Dilly J, Hoffman MT, Abbassi L, Li Z, Paradiso F, Parent BD, et al. Mechanisms of Resistance to Oncogenic KRAS Inhibition in Pancreatic Cancer. Cancer Discov. 2024;14(11):2135-2161.
[Google Scholar]
[CrossRef]
|
| 101. |
Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78-83.
[Google Scholar]
[CrossRef]
|
| 102. |
Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377.
[Google Scholar]
[CrossRef]
|
| 103. |
Shaurova T, Zhang L, Goodrich DW, Hershberger PA. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front Genet. 2020;11:281.
[Google Scholar]
[CrossRef]
|
| 104. |
Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388-2406.
[Google Scholar]
[CrossRef]
|
| 105. |
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427-1438.
[Google Scholar]
[CrossRef]
|
| 106. |
Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119(20):4597-4607.
[Google Scholar]
[CrossRef]
|
| 107. |
O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417-430.
[Google Scholar]
[CrossRef]
|
| 108. |
Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373:209-219.
[Google Scholar]
[CrossRef]
|
| 109. |
Morrison L, Loibl S, Turner NC. The CDK4/6 inhibitor revolution - a game-changing era for breast cancer treatment. Nat Rev Clin Oncol. 2024;21(2):89-105.
[Google Scholar]
[CrossRef]
|
| 110. |
Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425-439.
[Google Scholar]
[CrossRef]
|
| 111. |
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465-2472.
[Google Scholar]
[CrossRef]
|
| 112. |
Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol. 2017;35(32):3638-3646.
[Google Scholar]
[CrossRef]
|
| 113. |
Xue Y, Zhai J. Strategy of combining CDK4/6 inhibitors with other therapies and mechanisms of resistance. Int J Clin Exp Pathol. 2024;17(7):189-207.
[Google Scholar]
[CrossRef]
|
| 114. |
Rampioni Vinciguerra GL, Sonego M, Segatto I, Dall'Acqua A, Vecchione A, Baldassarre G, et al. CDK4/6 Inhibitors in Combination Therapies: Better in Company Than Alone: A Mini Review. Front Oncol. 2022;12:891580.
[Google Scholar]
[CrossRef]
|
| 115. |
Giugliano F, De Angelis C, Pistilli B, Viale G, Bianchini G, Giuliano M, et al. Overcoming Resistance to CDK4/6 inhibitors in Hormone Receptor positive, HER2 negative breast cancer: Innovative Combinations and Emerging Strategies. Cancer Treat Rev. 2025;139:102980.
[Google Scholar]
[CrossRef]
|
| 116. |
Liu Y, Park S, Li Y. Breaking Cancer's Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy. Cancers (Basel). 2025;17(12):1941.
[Google Scholar]
[CrossRef]
|
| 117. |
Goodwin CM, Waters AM, Klomp JE, Javaid S, Bryant KL, Stalnecker CA, et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer Res. 2023;83(1):141-157.
[Google Scholar]
[CrossRef]
|
| 118. |
Knudsen ES, Kumarasamy V, Ruiz A, Sivinski J, Chung S, Grant A, et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene. 2019.
[Google Scholar]
[CrossRef]
|
| 119. |
Kumarasamy V, Vail P, Nambiar R, Witkiewicz AK, Knudsen ES. Functional Determinants of Cell Cycle Plasticity and Sensitivity to CDK4/6 Inhibition. Cancer Res. 2021;81(5):1347-1360.
[Google Scholar]
[CrossRef]
|
| 120. |
Alvarez-Fernandez M, Malumbres M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell. 2020;37(4):514-529.
[Google Scholar]
[CrossRef]
|
| 121. |
Witkiewicz AK, Kumarasamy V, Sanidas I, Knudsen ES. Cancer cell cycle dystopia: heterogeneity, plasticity, and therapy. Trends Cancer. 2022;8(9):711-725.
[Google Scholar]
[CrossRef]
|
| 122. |
Suski JM, Braun M, Strmiska V, Sicinski P. Targeting cell-cycle machinery in cancer. Cancer Cell. 2021;39(6):759-778.
[Google Scholar]
[CrossRef]
|
| 123. |
Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362(6421):1416-1422.
[Google Scholar]
[CrossRef]
|
| 124. |
Ruscetti M, Morris JPt, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2020;181(2):424-441.e421.
[Google Scholar]
[CrossRef]
|
| 125. |
Knudsen ES, Kumarasamy V, Chung S, Ruiz A, Vail P, Tzetzo S, et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut. 2020;70(1):4-5.
[Google Scholar]
[CrossRef]
|
| 126. |
Kudo R, Safonov A, Jones C, Moiso E, Dry JR, Shao H, et al. Long-term breast cancer response to CDK4/6 inhibition defined by TP53-mediated geroconversion. Cancer Cell. 2024;42(11):1919-1935.e1919.
[Google Scholar]
[CrossRef]
|
| 127. |
Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568-576.
[Google Scholar]
[CrossRef]
|
| 128. |
Palmer CL, Boras B, Pascual B, Li N, Li D, Garza S, et al. CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety. Cancer Cell. 2025;43(3):464-481.e414.
[Google Scholar]
[CrossRef]
|
| 129. |
Arora M, Moser J, Hoffman TE, Watts LP, Min M, Musteanu M, et al. Rapid adaptation to CDK2 inhibition exposes intrinsic cell-cycle plasticity. Cell. 2023;186(12):2628-2643.e2621.
[Google Scholar]
[CrossRef]
|

 ,
Thomas N. O’Connor
,
Thomas N. O’Connor