Abstract
Cancer stem cells (CSCs) represent a subpopulation of cancer cells characterized by their capacity for self-renewal, differentiation, and tumorigenicity. CSCs exist along a spectrum of stemness regulated by both intrinsic factors and extrinsic signals from the tumor microenvironment (TME). The TME is composed of diverse cell types such as stromal and immune cells, and also physical factors such as the extracellular matrix and hypoxia. Environmental signals originating from the TME can induce non-CSCs to acquire stem-like traits, while CSCs in turn modulate the TME by recruiting and reprogramming immune and stromal cells. Analogous to normal stem cell niches, CSCs reside in or construct supportive niches that promote stemness, metastasis, immune evasion, and therapy resistance. This reciprocal interaction between CSCs and the TME underscores the complexity of cancer stemness and presents challenges and opportunities for therapeutics.
Keywords
Cancer stem cells, tumor microenvironment, cellular plasticity, CSC niche
1. Introduction
Cancer stem cells (CSCs) are a subset of cells within the heterogeneous population of tumoral cells that are distinguished by their ability to self-renew and differentiate into other cancer cell subpopulations [1]. The concept of CSCs originally arose from the identification of cancer cell subpopulations exhibiting highly tumorigenic properties [2,3]. Early models of CSCs leaned toward a discrete, binary classification: cancer cells either were non-CSCs or CSCs, with CSCs defined by the ability to self-renew, differentiate, and divide either symmetrically or asymmetrically [4]. Further work characterizing the other properties of CSCs, such as drug resistance and metastatic potential [5,6], combined with identification of the pathways contributing to CSC maintenance, such as Wnt, Notch, and Hedgehog signaling [7], has led to the refinement and development of new frameworks for understanding CSCs. Within tumors, cancer cells are now widely understood to lie along a dynamic spectrum ranging from highly differentiated cells to those exhibiting varying levels of stemness [8]. Like normal human stem cells, CSC stemness is controlled by a highly complex network of environmental signals, where non-CSCs can be directed to take on a more stem-like phenotype and vice versa [9]. Accordingly, methods for evaluating cancer cell stemness involve in vitro assessment of sphere/colony formation, stemness markers such as SOX2, Nanog, and Oct4, and in vivo tumorigenesis [10]. Cell sorting techniques to evaluate aldehyde dehydrogenase (ALDH) activity and cell surface markers such as CD44 and CD133 are also used to determine and isolate CSCs, but specific CSC marker sets vary by cancer type and may also be expressed by non-cancer cells [11].
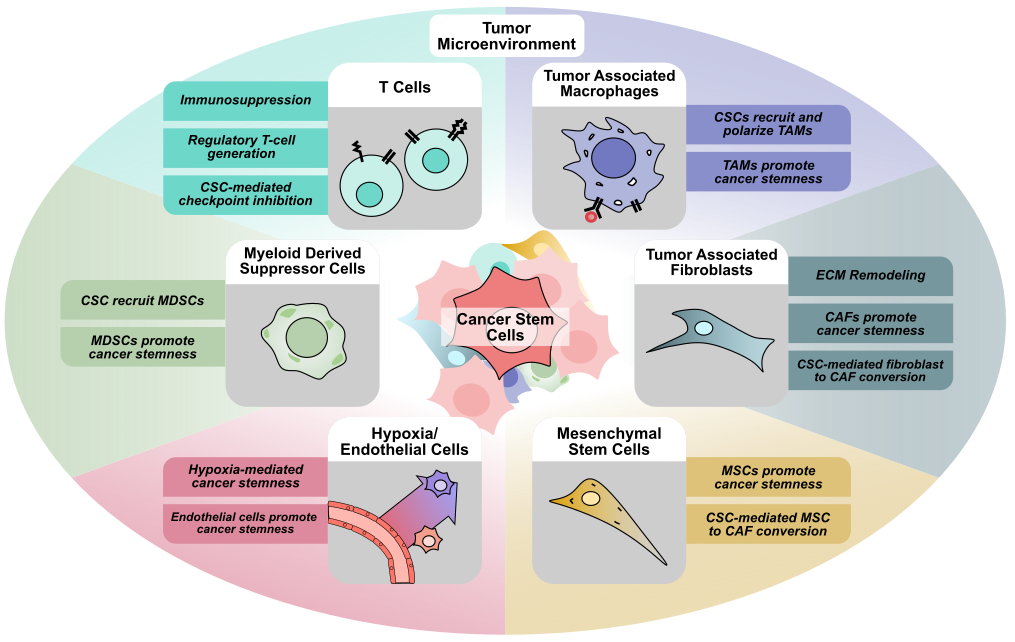
Within tumors, cancer cells and CSCs do not exist in isolation, and are part of a complex network known as the tumor microenvironment (TME). The tumor microenvironment consists of a heterogeneous population of cells, including cancer, immune, and stromal cells, as well as other cells that make up larger structures within the TME such as blood vessels and nerves [12]. Initially thought to only play a passive role in tumor progression, these non-tumoral cells residing within the TME have been implicated in nearly all aspects of tumor biology (Figure 1). Numerous cell types such as fibroblasts, also known as cancer-associated fibroblasts, endothelial cells, mesenchymal stem cells, adipocytes, neurons, and immune cells drive tumor initiation, proliferation, metastasis, and cancer cell stemness [12]. Additionally, many immune cell types, such as CD8+ or CD4+ T cells, natural killer cells, dendritic cells, regulatory T cells, myeloid-derived suppressor cells, and macrophages have also been demonstrated to play an active role within the tumor microenvironment. Interactions between cancer cells and these immune cells also contribute to cancer stem cell maintenance, immune cell reprogramming, immune suppression, and are the basis for cancer immunotherapy [13]. Beyond its cellular components, the TME also encompasses the unique physiological conditions present within each tumor, such as hypoxia, low pH, and extracellular matrix stiffness [14]. Within the TME, the effects of different conditions combined with complex networks of signaling between the different cell types have been shown to regulate key features of cancer such as tumor proliferation, metastasis, and treatment resistance [15].
2. Cancer-Associated Fibroblasts and CSC Maintenance
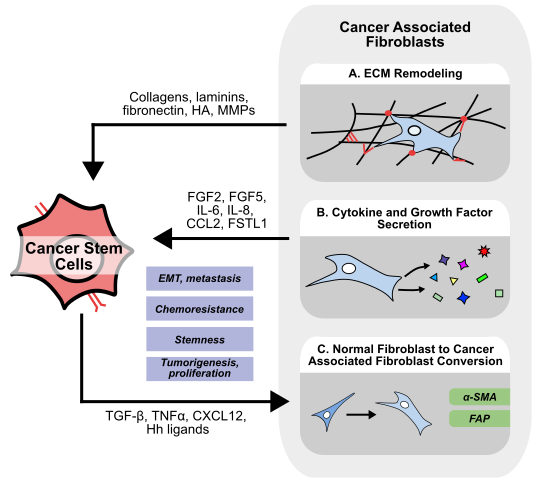
Found throughout the body, fibroblasts play a critical role in tissue repair, stroma homeostasis, and have been implicated in several diseases, including cancer [16]. To meet the diverse needs of the specialized tissues they reside in, fibroblasts display large inter-organ heterogeneity [17], but recent advances have also demonstrated a significant intra-organ and intra-tumor heterogeneity of fibroblasts [18,19]. Fibroblasts are well known for their ability to synthesize and maintain the extracellular matrix (ECM), and to secrete a variety of growth factors, cytokines, and chemokines to regulate other tissue-resident cells and processes (Figure 2) [16]. Within the tumor microenvironment, cancer-associated fibroblasts (CAFs) could be simply defined as fibroblast-like cells in close proximity to other cancer cells. However, in reality, distinguishing between normal fibroblasts and CAFs poses a challenge. Typically, CAFs are distinguished from normal fibroblasts by expression of alpha smooth muscle actin (α-SMA) and fibroblast activation protein (FAP), and display an altered secretome involving molecules such as IL-6 and CCL2 [20]. However, across different types of cancers, there are numerous suggested origins and mechanisms of activation for CAFs [21]. While many studies have shown that IL-1 and transforming growth factor-beta (TGF-β) are responsible for normal fibroblast to CAF transformation [22], CAFs may also originate from other cells such as mesenchymal stem cells.
2.1 CAF-mediated ECM remodeling
Within the tumor microenvironment, fibroblasts are the predominant regulator of the ECM, and are responsible for secreting, crosslinking, and remodeling structural proteins such as collagens, fibronectin, and laminins (Figure 2A) [23]. Apart from providing structure and stability within the TME, these structural molecules can bind many types of extracellular-sensing receptors on cancer cells to regulate their stemness [24]. For example, CAFs are known to secrete laminins such as laminin-332, which induces a stem-like phenotype in hepatocellular carcinoma (HCC), promoting quiescence and chemoresistance in an mTORC2-dependent manner [25,26]. Other laminins interact with integrins, such as integrin α6β4, on cancer cells to activate downstream Notch signaling, and promote cell proliferation and migration [27]. Similarly, activation of integrin ITGA2 on cancer cells upon interaction with collagen I promotes tumorigenesis and sphere formation in oral squamous cell carcinoma (OSCC) [28].
Other ECM components, such as fibronectin, interact with integrin αvβ3 on cancer cells to activate PI3K/Akt/SOX2 and CDC42/YAP-1/NUPR1/Nestin axes, promoting a stem-like phenotype in glioblastoma (GBM) [29]. Similarly, Wu et al. demonstrate that CAF-secreted fibronectin activates PI3K/Akt/SOX2 signaling in non-small cell lung cancer (NSCLC), and that inhibition of αvβ3 can sensitize cancer cells to chemotherapy [30]. Fibronectin also activates Wnt/β-catenin and MAPK/ERK signaling within NSCLC cells to promote angiogenesis and invasion in a WISP3-dependent manner [31]. Additionally, fibronectin-integrin interaction can upregulate the epithelial-mesenchymal transition (EMT) markers snail, N-cadherin, and vimentin [32], while fibronectin can promote cell migration, invasion, and matrix metalloproteinase 2 (MMP-2) expression in breast cancer [33]. CAF-derived MMPs are responsible for remodeling tumoral extracellular matrix, promoting angiogenesis, and degrading basement membrane to help promote EMT and invasion [34,35]. Given that MMP-7 has been shown to promote chemoresistance in numerous types of CSCs by mediating MUC-1 shedding [36], CAF-derived MMPs may also play a role in regulating stemness independent of canonical MMP-EMT pathways.
CAFs have been shown to secrete other structural molecules such as the polysaccharide hyaluronic acid (HA) in response to TGF-β [37]. In breast cancer, HA activates the stemness-related pathways Ras-MAPK and PI3K/Akt by binding to CD44, a widely acknowledged marker for CSCs [38,39]. Similarly, in head and neck squamous cell carcinoma (HNSCC), HA/CD44 mediated signaling enhances SOX2 expression through the PI3K/4EBP1 axis [40]. Apart from secreting structural proteins, CAFs can also regulate ECM stiffness by lysyl oxidase-mediated crosslinking of the existing ECM [41]. In HCC, matrix stiffening in 3D hydrogel culture is associated with increased expression of stemness markers CD44, EpCAM, and Nanog [42], while matrix stiffening promotes EMT in lung cancer in a DDR2-dependent manner [43]. Similarly, in colorectal cancer (CRC) cells, matrix stiffness is associated with increased CSC markers like CD133, ALDH1, and LGR-5, and knockdown of the Hippo transcription factor, Yes-associated protein (YAP), could reverse this effect and reduce sphere formation in vitro [44]. Together, these findings suggest that the ECM serves as more than a passive scaffold within the TME; the ECM is dynamically deposited and remodeled by CAFs to regulate cancer cell stemness via structural proteins and ECM-sensing receptors.
2.2 CAF-secreted growth factors
Apart from regulating the ECM, fibroblasts also secrete a variety of soluble factors to influence cancer cell migration, proliferation, and stemness (Figure 2B) [45]. Breast cancer-associated fibroblasts secrete fibroblast growth factor 5 (FGF5) to upregulate stemness markers ID3 and SOX10, and enhance sphere formation in vitro [46]. Similarly, breast cancer CAFs express high FGF2, and CAF-derived conditioned media promotes cancer cell proliferation in vivo, while knockdown of FGF2 in CAFs abrogated this effect [47]. Mechanistically, CAF FGF2 secretion is activated by estrogen via GPER/EGFR/ERK signaling [48]. Given that FGF2 helps maintain an undifferentiated state in mesenchymal stem cells marked by high expression of Oct4, SOX2, and Nanog [49], is also reported to convert iPSCs into CSCs, and is used in spheroid culture to enrich CSC populations [50,51], CAF-derived FGF2 may also play a larger role in regulating other cells within the TME.
Unsurprisingly, cancer cell and CAF communication is bidirectional. Cancer cells can secrete hedgehog ligands to promote FGF5 secretion from CAFs [46], while cancer cell-derived TGF-β can activate normal fibroblasts to adopt a more CAF-like phenotype (Figure 2C) [52]. In breast cancer, cancer cells secrete tumor necrosis factor α (TNFα) to induce CAFs secretion of hepatocyte growth factor (HGF), which promotes cancer cell proliferation, EMT, and radioresistance in vitro. Interestingly, radiation-exposed breast cancer cells secrete more TNFα to further enhance radioresistance in a CAF-dependent manner [53]. In gastric cancer, CAF-derived HGF promotes cell proliferation and migration in only Met-unamplified cells, while CAF-derived HGF enriched the proportion of CD44+ CRC stem cells in vitro and promoted metastasis through increased adhesion to endothelial cells in vivo [54,55]. In HCC, both fibroblast and cancer cell-derived TGF-β enhance expression of FSTL1 within CAFs. CAF-derived FSTL1 promotes tumorigenesis, metastasis, and sorafenib resistance in vivo [56]. In breast cancer, FSTL1 promotes stemness markers, sphere formation, and chemoresistance in vitro, while also promoting tumorigenesis and metastasis in gastric cancer [57,58]. In HCC, spatial multiomics identifies a subset of CAFs marked by high expression of FSTL1 that are localized within regions of cancer stemness. Unsurprisingly, these regions demonstrate enrichment of TGF-β and Notch signaling [59].
2.3 CAF-derived cytokines and chemokines
In gastric cancer, CAFs secrete IL-6 to promote EMT and metastasis by activating the JAK/STAT3 axis, while IL-6 promotes sphere formation and the expression of stemness markers Oct4, SOX2, CD44, and Nanog in HCC [60,61]. Similarly, breast cancer CAFs secrete IL-6 to promote radioresistance via STAT3 signaling, while CAF-derived IL-6 enhances p53 ubiquitination and degradation, allowing cells to escape doxorubicin-induced apoptosis [62,63]. High levels of other CAF-derived cytokines such as IL-8 are associated with poor response to chemotherapy and cisplatin resistance in gastric cancer [64]. In lung cancer, IL-8-secreting CAFs are marked by high levels of the filament Desmin, and are activated by cancer cell-derived lactate. Normal fibroblasts co-injected with lung cancer cells enhanced tumor growth in vivo, while knockout of the Desmin transcription factor JUNB-B in normal fibroblasts attenuated this effect [65]. Similarly, in ovarian cancer, CAF-secreted IL-8 promotes sphere formation and stemness in vitro, while CAF co-injection with ovarian cancer cells promotes tumor growth in vivo [66]. In OSCC, CAFs also produce the chemokine CXCL12 to increase sphere formation and expression of stemness markers in cancer cells [67]. CXCL12 has also been shown to stimulate normal fibroblasts to adopt CAF-like properties in vitro [68], suggesting that CAF secretion of CXCL12 may have a positive feedback effect. Given that CXCL12/CXCR4 signaling activates oncogenic ERK and PI3K/Akt signaling, and promotes chemotherapy resistance [69], it is unsurprising that high expression of CXCL2 is associated with metastasis to the brain, bone, lung, and liver in breast cancer [70]. Additionally, there are a number of other CAF-derived cytokines that regulate CSCs such as CAF-derived CCL2, which promotes CSC self-renewal via NOTCH1 signaling in breast cancer [71].
3. Immune Cells
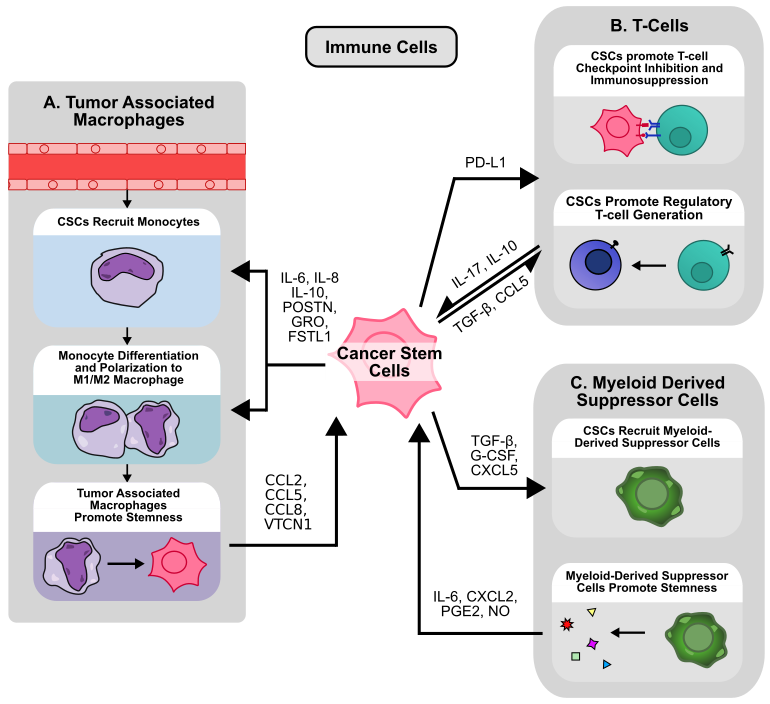
Apart from CAFs, endothelial cells, and cancer cells, the tumor microenvironment also contains a heterogeneous population of immune cells [72]. At the most basic level, these immune cells can be classified either as tumor-promoting or tumor-suppressing [13]. CD8+ and CD4+ T cells are widely known for their ability to target and eliminate cancer cells [73], while other cells like natural killer (NK) cells, dendritic cells, and M1 macrophages also play a critical role in identifying and eliminating cancer cells within the TME [13]. On the other hand, regulatory T cells (Tregs), myeloid-derived suppressive cells (MDSCs), and M2 macrophages are recognized to be tumor-promoting, either by modulating other immune cells or interacting directly with cancer cells [13,74]. Unsurprisingly, within the TME, cancer cells and CSCs are not bystanders to immune cell activity. CSCs actively secrete signaling molecules to attract and modulate immune cell activity, and are likewise influenced by immune cell signaling. Moreover, CSCs also play an important role in escaping immune surveillance and inhibiting immune response in the TME (Figure 3) [75].
3.1 Tumor associated macrophages
Tumor associated macrophages (TAMs) are macrophages found within the TME, and originate either from tissue resident macrophages or circulating monocytes, the latter of which is the main source of TAM replenishment [76]. Monocyte-to-macrophage differentiation is driven by complex environmental cues and immune/cancer cell secreted cytokines such as colony stimulating factor (CSF) 1, CSF-2, and IL-34 [77]. Once differentiated, these monocytes can be further polarized by signals within the TME into two general categories, M1-polarized and M2-polarized macrophages. M1-polarized macrophages are recognized as tumor-suppressive, while M2-polarized macrophages are known for their tumor-promoting abilities [78]. However, these categories are likely an oversimplification, and macrophages in vivo exist on a spectrum of activation states influenced by a complex network of immune signals [79], of which CSCs play a critical role (Figure 3A). For example, in GBM, CSCs preferentially express periostin (POSTN) to recruit monocytes to the tumor site and promote polarization to an M2, tumor-promoting state via integrin αvβ3 signaling, a finding recapitulated in ICC [80,81]. Similarly, glioma stem cells (GSCs) induced STAT3 phosphorylation and STAT1 dephosphorylation in macrophages to promote M2-polarization and secretion of immunosuppressive IL-10 [82]. Inflammatory breast CSC-like cells also recruit monocytes and promote M2-polarization via IL-8 and GRO-mediated STAT3 phosphorylation in monocytes [83]. Additionally, numerous other studies have demonstrated the ability for CSCs to polarize monocytes to M2-like macrophages [84,85]. CD133+ GSC-derived IL-6 and IL-10 activate STAT3 signaling within macrophages to activate downstream expression of VTCN1. VTCN1 inhibits macrophage phagocytosis, T cell IL-2/IFN-γ production, and promotes T cell apoptosis, and silencing of VTCN1 in macrophages reduces tumor growth in vivo [86]. GSCs also preferentially express WISP1, which promotes stemness in an autocrine manner while promoting M2 TAM survival via integrin α6β1/Akt signaling [87]. Likewise, GSC-derived FSTL1 promotes stemness by activating PI3K-Akt signaling in an autocrine manner, while also promoting macrophage recruitment and M2 polarization in vivo [88].
CD14+ M2-like macrophages enhance sphere formation, stemness marker expression in GBM cells, and enrich the population of CD133+/CD44+ CSCs via CCL2 secretion. Co-injection of these M2-like macrophages with cancer cells enhances tumor growth in vivo, an effect reversed by inhibition of CCR2 [84]. Similarly, TAM-derived CCL8 promotes stemness of GBM cells cultured in vitro, while promoting tumor growth in vivo. Interestingly, cells cultured under sphere conditions overexpressed the CCL8 receptors CCR1 and CCR5 [89]. TAM-derived cytokines are also implicated in CSC regulation in numerous other cancer types. In breast cancer, high infiltration of CD163+ M2 macrophages in the TME is associated with lymph node metastasis and decreased survival, and TAM-derived CCL2 is responsible for enhancing the proportion of CD24-/CD44+ or ALDH+ CSCs via CCL2/Akt/β-catenin signaling in vitro [90]. Additionally, TAM-derived IL-6 promotes stemness via STAT3 signaling in HCC cells, and TAM-derived CCL5 similarly promotes stemness via β-catenin/STAT3 signaling in prostate cancer. Knockdown of CCL5 in TAMs inhibited tumor growth, metastasis, and CSC populations in vivo [91,92]. In HCC, spatial transcriptomic analysis suggests that CSCs are colocalized with SPP1+ macrophages within the TME, which are typically regarded as tumor-promoting M2-like macrophages [93,94]. Consistent with their role in tumor progression, SPP1+ macrophage and CSC colocalization is associated with poorer survival in patients with HCC. Interestingly, Fan et al. also show enhanced HIF-1α expression within colocalized areas [93]. Accordingly, HCC cells have been shown to secrete miR-1290 to induce M2-macrophage polarization [95]. Moreover, in HCC, SPP1+ macrophages are frequently found in close proximity to FAP+ CAFs [96,97], suggesting that macrophages, CAFs, and CSCs may form a complex network to regulate stemness within the TME. Given that M2 macrophages also suppress immune response in HCC by promoting the expression of exhaustion markers PD1/TIM3 on CD8+ T cells [98], CSCs may also modulate immune response via M2 macrophages.
While M1-macrophages, as the counterpart to M2-macrophages, were originally classified as tumor-suppressive, high levels of iNOS+ M1-polarized macrophages have been suggested to be associated with improved survival in HER2+ breast cancer [99]. However, Oshi et al. demonstrate that upon higher dimensional, transcriptomic classification of M1/M2 macrophages, M1 or the ratio of M1/M2 macrophages are not associated with improved survival in breast cancer [100], suggesting that the role of M1 macrophages may be more complex. In vitro, M1-polarized macrophages enrich the populations of CD24-/CD44+ and ALDH1+ breast CSCs during co-culture, while increasing the expression of stemness markers Lin-28B and Nanog in breast cancer cells [101]. Similarly, conditioned media from LPS/IFNγ-induced M1 macrophages enhances the expression of stemness markers in breast cancer cells via NFκB signaling [102]. In CRC, ID1-expressing M1-like macrophages have pro-tumorigenic and stemness promoting properties. Tumors grown in myeloid-specific ID1 knockout mice contain fewer ALDH+ and CD44+/Lgfr5+ CSCs, and inhibition of ID1 deubiquitination in M1 macrophages with ML323 reduces tumor growth, metastasis, and sensitizes tumors to chemo/immunotherapy [103]. These findings suggest that M1 macrophages, traditionally classified as tumor-suppressing, can also influence CSC maintenance and promote tumor growth.
3.2 CSCs and checkpoint inhibition
In addition to macrophages, CSCs also interact with other immune cells in the TME such as CD8+ and CD4+ T cells (Figure 3B). While activated CD8+ T cells are highly effective in clearing cancer cells, exhausted CD8+ T cells exhibit diminished effector function marked by high expression of checkpoint inhibitor receptors such as PD-1, CTLA-4, and TIM-3 [104]. While immunotherapies targeting these pathways can restore immune cell activity and enhance tumor clearance [105], CSCs have been shown to undermine these responses by mediating the expression of checkpoint inhibitors and activating additional mechanisms of immune evasion. For example, immunohistochemistry (IHC) analysis of CD44 expression, a common stem cell marker in lung adenocarcinoma (LUAD), reveals a positive association with PD-L1 expression [106], while CD133+/CD44+ CRC stem cells likewise exhibit higher PD-L1 expression via enhanced STAT3 signaling in patient-derived xenograft (PDX) models [107]. In breast cancer, CD24-/CD44+/EpCAM+ CSCs express higher levels of PD-L1 via Notch3/mTOR signaling in vitro [108], while ALDH1A1 expression by IHC in breast cancer is also associated with increased PD-L1 levels [109]. However, these associations may be context/cancer dependent. IHC analysis in NSCLC demonstrates a negative correlation between ALDH1 and PD-L1, and no significant relationship between CD44 and PD-L1 expression [110], suggesting that the relationship between stemness and PD-L1 expression may be more complex. Moreover, TGF-β-induced EMT increased PD-L1 levels in breast CSCs via β-catenin/STT3 glycosylation and stabilization of PD-L1 in vitro [111]. In HNSCC, IFN-I/IFNAR1 signaling promotes stemness and production of exosomal PD-L1 and Galectin-9, while IFNAR knockdown reduces the proportion of CD44+/ALDH+ CSCs, inhibits tumor growth, and promotes T-cell exhaustion in vivo [112]. However, CSC-derived PD-L1 not only acts on immune cells, but also on other cancer cells. In CRC, CD133+/CD44+ CSCs express higher levels of PD-L1, which interacts with HMGA1 to enhance sphere formation and Oct4 expression via Akt and MEK signaling [113], suggesting that CSCs can induce stemness within other cells in addition to suppressing immune response via PD-L1 expression.
3.3 Other immune cells
Apart from regulating CD8+ cells, CSCs also influence the activation and function of various other numerous immune cell populations. In breast cancer, CSCs promote NK cell migration but inhibit NK cell-mediated cytotoxicity. These CSCs express higher levels of NK-inhibiting ligand HLA-E, while downregulating the NK-activating ligands MICA and MICB [114]. However, other studies suggest that NK cells preferentially target CSCs in various cancers including breast cancer, glioma, sarcoma, and pancreatic cancer [115]. Additionally, CD4+ T cells cultured in the presence of breast CSC-conditioned media contain a larger proportion of CD4+CD25+CD127− Tregs, possibly via CSC-derived TGF-β (Figure 3B) [116]. Breast CSCs also secrete exosomal FOXP3 to transform CD4+ T cells into CD4+/CD25+/FOXP3+ Tregs in vitro [117]. In GBM, CSCs not only promote the generation of Tregs, but also inhibit CD4+ and CD8+ cell activation and proliferation via activation of STAT3 signaling in T cells [118]. In HNSCC, CD44+ CSCs produce higher levels of numerous cytokines including TGF- β, and also promote Treg generation, MDSC generation, and inhibit T cell activation from PBMCs [119]. Ovarian CSCs not only induced Tregs to produce higher levels of the immunosuppressive cytokine IL-10, but also promoted the recruitment and chemotaxis of Tregs in vitro via CCL5 secretion [120]. In vivo, ovarian CD133+ CSCs express higher levels of the IL-17 receptor compared to non-CSCs, and colocalize with IL-17-producing cells such as CD4+ TH17 cells and macrophages. This IL-17 promotes sphere formation in vitro and tumorigenesis in vivo via NF-κB and p38 MAPK signaling in cancer cells [121]. Similarly, IL-17 promotes stemness in pancreatic cancer cells via NF-κB signaling [122].
3.4 Myeloid-derived suppressor cells
Other important immunosuppressive cells in the TME include MDSCs. MDSCs are a heterogeneous population of cells broadly classified for their immunosuppressive ability. MDSCs arise from myeloid progenitors in the setting of abnormal maturation and development, often due to cancer, chronic inflammation, or infection [123]. MDSCs are typically classified into two categories, polymorphonuclear and monocytic MDSCs (PMN-MDSC and M-MDSC). Although some typical surface marker sets are used to distinguish the two, higher dimension analysis has revealed many subpopulations within each category [124]. In the setting of cancer, MDSCs are recruited to the TME by chemokines such as CCL2 and CXCL12, where they can activate and proliferate via stimulation of STAT3, STAT6, and NF-κB pathways, among many others [125]. In breast cancer, CSCs preferentially express G-CSF via enhanced mTOR signaling, which results in MDSC accumulation within the TME [126], while melanoma CSC-derived TGF-β similarly recruits PMN-MDSCs to the tumor site [127]. In prostate cancer, Yap1-high CSC-like cells express higher levels of CXCL5, which promotes MDSC recruitment via CXCR2 signaling [128] (Figure 3C).
MDSCs are well-established mediators of T-cell immunosuppression, employing mechanisms such as L-arginine depletion, nitric oxide (NO) production, and reactive oxygen species (ROS) generation [129], but also regulate cancer cell stemness within the TME. In breast cancer, PMN-MDSCs enrich the proportion of ALDH+ CSCs, increase sphere formation, and upregulate expression of stemness markers via CXCL2/CXCR2 signaling [130]. Similarly, MDSCs cocultured with ovarian cancer cells enhance sphere formation ability and stemness markers via CSF-2/STAT3 signaling [131]. MDSC-derived IL-6 and NO activate STAT3 and NOTCH signaling pathways in vitro to enrich the population of ALDH+ CSCs in breast cancer [132]. Given that inducible nitric oxide synthase (iNOS) expression and subsequent NO production within cancer cells are associated with CSC maintenance via Notch1 signaling in numerous types of cancers [133,134], it is possible that MDSC-derived NO may exert a similar effect in cancers other than breast cancer. Similarly, while few studies have examined the role of MDSC-derived ROS in relation to CSCs, cancer cell-derived ROS are known to play multiple important roles in CSC maintenance, including ROS-mediated HIF stabilization [135]. In ovarian cancer, tumor-derived G-CSF promotes the generation and survival of MDSCs in vivo [136]. These MDSCs, in turn, secrete prostaglandin E2 (PGE2) to increase stemness and PD-L1 expression via the PI3K/Akt axis. Accordingly, G-CSF overexpressing tumors contain elevated levels of ALDH+ CSCs in vivo [136,137]. Additionally, in a mouse breast cancer model, PNM-MDSCs maintain the CD49f+ mouse CSC population via netrin-1 signaling. Notably, while anti-CTLA-4 immunotherapy enriched the population of CD49f+ CSCs, combination with netrin-1 blockade abolished this effect [138], suggesting that MDSC-mediated regulation of stemness may also contribute to therapy-induced CSC expansion.
4. Mesenchymal Stem Cells
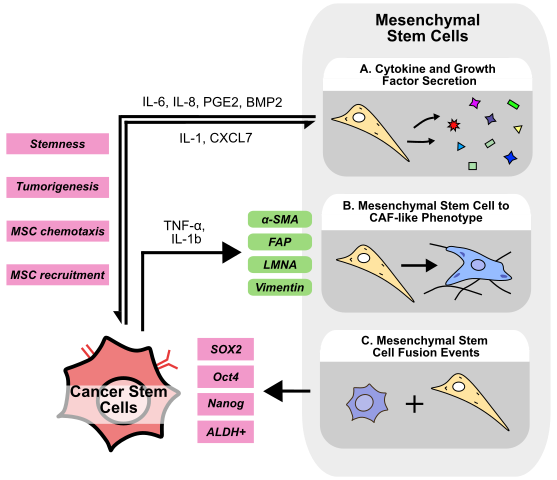
Mesenchymal stem cells (MSCs) are multipotent stromal cells responsible for tissue repair, homeostasis, and immune response modulation. MSCs have been shown to differentiate into many cell types including adipocytes, osteocytes, and chondrocytes [139], and are present and able to be isolated from numerous types of tissue including bone marrow, adipose, placental, and umbilical cord tissue [140]. Isolated MSCs are typically characterized with markers such as CD105+/CD73+/CD90+ and CD45-, CD34-, MHC-II-, but MSCs are highly heterogenous and show variation both between and within tissue of origin [141]. Regardless, numerous clinical trials are investigating MSC isolation, expansion, and transplantation for a wide range of diseases [142]. MSCs secrete a variety of growth factors, cytokines, and miRNAs to promote tissue repair; however, these same molecules have also been implicated in cancer development and progression [143]. For example, MSC-derived TGF-β and PGE2 have been demonstrated to inhibit CD8+ T, NK, and dendritic cell-mediated inflammatory responses to cancer cells, and also to promote tumor-promoting M2-macrophage polarization [144]. Beyond immunosuppression, MSCs also play a pivotal role in mediating cancer cell stemness within the TME (Figure 4).
4.1 MSCs-secreted cytokines and chemokines
Numerous cytokines secreted by MSCs have been shown to promote stemness (Figure 4A). MSCs express higher levels of BMP2 upon coculture with ovarian cancer cells, which acts in a paracrine manner to increase the number of ALDH+/CD133+ CSCs and enhance sphere formation ability in vitro, an effect abolished upon treatment with the BMP signaling inhibitor, Noggin [145]. Furthermore, primary MSCs co-cultured with GSCs promote sphere formation in vitro, and tumorigenesis in vivo via MSC-derived IL-6/STAT3 signaling [146]. Similarly, MSCs isolated from human bone marrow (BM-MSCs) secrete IL-6 to promote cell invasion in HCC [147]. IL-6 has also been shown to enrich CSC populations and promote treatment resistance in several cancers including breast cancer, HNSCC, and colorectal cancer [148–150], suggesting that MSC-derived IL-6 may play a central role in promoting stemness across cancer types. In breast cancer, ALDH+ MSCs secrete CXCL7 to induce cancer cell IL-6 secretion, which can then promote MSC chemotaxis in a positive feedback loop. Accordingly, ALDH+ MSCs injected into the mouse tibia were targeted to the breast tumor site, promoting tumor growth and enriching the proportion of ALDH+ or CD24-/CD44+ CSCs in vivo [151]. Additionally, while MSCs are recognized to secrete PGE2 to promote recovery after liver injury [152], MSC-derived PGE2 also mediates CSC maintenance. When co-cultured with MSCs, CRC cells secrete IL-1, which stimulates MSCs to produce PGE2, IL-6, and IL-8. Co-injection of MSCs with CRC cells promotes tumorigenesis and enriches the ALDH+ CSC population via PGE2/Akt/β-catenin signaling [153]. PGE2 has also been demonstrated to promote cancer cell stemness in lung cancer via PI3K/Akt signaling, suggesting the effects of MSC-derived PGE2 may not be restricted to only CRC [154]. Similar to the findings reported by Liu et al. in breast cancer, BM-MSCs injected into the bloodstream are recruited into mouse prostate tumors, which then increase the number of CD44+/CD133+ CSCs. In vitro, co-culture of BM-MSCs with prostate cancer cells promotes sphere formation and the expression of stemness markers via downregulation of AR signaling [155]. In GBM mouse models, co-injection of MSCs with GBM cells likewise enhances tumor growth and enriches the population of tumoral CSCs [156]. In HCC, tumor-specific CD34+/CLDN5+ endothelial cell-derived IGF can recruit MSCs into the TME, which induces a stem-like phenotype in HCC cells [157], further supporting the notion that cells within the TME can actively recruit MSCs to enhance cancer cell stemness.
4.2 Other roles of MSCS
Apart from directly mediating cancer stemness, MSCs have also been suggested to convert to a CAF-like phenotype (Figure 4B). Long-term stimulation with TNF-α and IL-1b imparts MSCs with CAF-like morphology, increased vimentin and FAP expression, and an increased ability to contract collagen gels [158]. MSCs cultured in stiff, ECM-mimicking conditions acquire a CAF-like phenotype upon treatment with breast cancer cell-derived conditioned media. These CAF-like cells downregulate the MSC marker Stro-1 while upregulating CAF markers α-SMA and LMNA [159]. Similarly, transcriptomic analysis of MSCs treated with exosomes derived from colorectal cancer cells reveals an increase in CAF-related genes such as α-SMA and FAP [160]. Considering the substantial evidence supporting the role of CAFs in promoting stemness, context-dependent transformation of MSCs to CAFs may represent an additional mechanism of MSC-mediated enhancement of cancer stemness. Additionally, MSCs may also fuse with non-CSCs to generate CSC-like cells (Figure 4C). Dornin et al. provide evidence to suggest that breast cancer cells fused with MSCs contain a higher proportion of ALDH+ CSCs and also express higher EMT markers [161]. Similarly, lung cancer-MSC and normal epithelial cell GES1-MSC fusion hybrids express higher levels of EMT markers and possess increased tumorigenicity in vivo [162,163]. Fusion hybrids of GBM cells and MSCs demonstrate higher stemness markers SOX2, Oct4, and Nanog in vitro, while displaying increased tumorigenicity in vivo [164].
5. Exosomal Signaling and Stemness
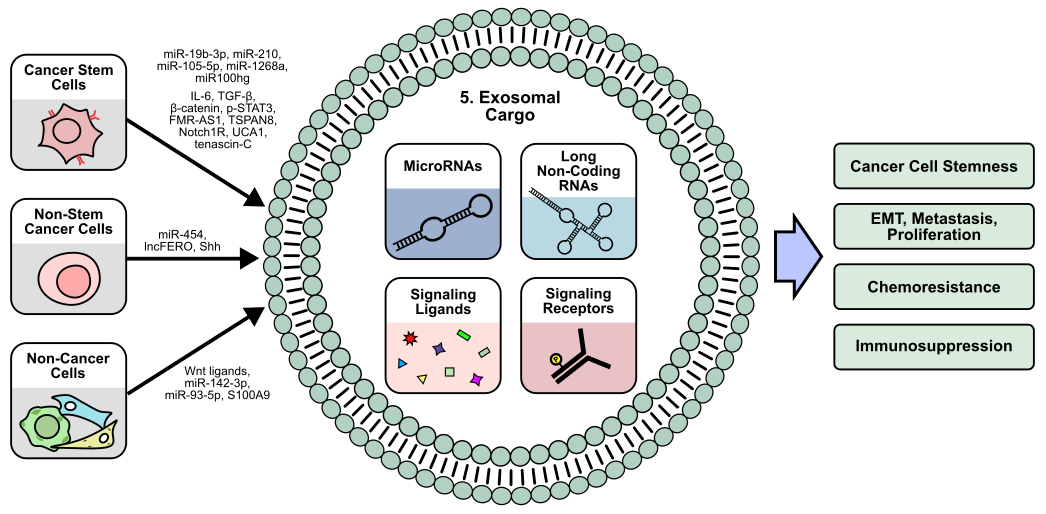
While a substantial portion of cellular signaling is mediated by small, soluble factors such as cytokines and chemokines, exosomes facilitate the transfer of larger macromolecules and insoluble factors, such as RNA, DNA, lipids, and proteins [165]. Exosomes are extracellular vesicles generated from the endocytosis of the plasma membrane, and thereby contain cell surface proteins that function to mediate their uptake and release [166]. Exosome formation is highly regulated and classified into ESCRT-dependent and independent pathways, which each consist of complex cascades governing cargo selection, membrane manipulation, and eventual exosome release [167]. As an important method of cell-to-cell communication, exosomes are critical to normal human development and cellular function, while exosomal dysregulation has been implicated in a wide variety of diseases including cancer [168]. Within the TME, exosomes and exosomal dysregulation have been shown to contribute to tumor proliferation, metastasis, and immune suppression (Figure 5) [169].
5.1 Cancer cell-derived exosomes
Additionally, exosomes have also been shown to promote stemness in recipient cells. For example, pancreatic ductal adenocarcinoma (PDAC) CSCs secrete exosomes enriched in tetraspanin 8 (TSPAN8) compared to their non-CSC counterparts. Exosomal TSPAN8 activates Hedgehog (Hh) signaling in recipient cells to promote colony formation, stemness marker expression, and tumorigenesis [170]. Similarly, in HCC, non-CSC-derived exosomes containing the Hh ligand Sonic hedgehog (Shh) could enhance recipient cell stemness [171]. In esophageal squamous cell carcinoma, CD44+ CSCs secrete exosomes containing the lncRNA FMR-AS1, which acts as a TLR7 ligand to promote TLR7/NF-κB mediated stemness in cancer cells [172]. In GBM, CSC-derived exosomes containing the Notch1 receptor enhance sphere formation, stemness marker expression, and tumorigenesis in recipient cells [173], suggesting that exosome-mediated stemness may not only be restricted to the transfer of ligands, but functional receptors as well.
In addition to exosomal proteins, exosomal RNA has also been widely studied in the context of CSCs. In breast cancer, exosomal miR-105-5p produced by CSCs promotes cancer cell migration and tumor growth by inhibiting the translation of GPR12 [174], while exosomal miR-454 derived from breast cancer non-CSCs activates Wnt signaling to expand the population of CD44+/CD133+ CSCs in vitro while enhancing tumor growth in vivo [175]. In melanoma, CSC-derived exosomes containing miR-1268a and miRNA-4535 have been shown to promote metastasis in non-CSCs by inhibiting autophagy [176,177], while lung CSCs secrete miR-210-3p via exosomes to promote EMT by targeting fibroblast growth factor receptor 1 (FGFRL1) translation in recipient cancer cells [178]. Similarly, in renal cell carcinoma (RCC), CSCs produce exosomal miR-19b-3p to promote EMT in recipient cells by inhibiting PTEN translation. Interestingly, these RCC CSC-derived exosomes have differential effects on tumor growth based on size and CD103+ incorporation, with high CD103+ exosomes being able to more efficiently target tumor cells [179]. miRNAs are known to be regulated by lncRNAs, which can sponge miRNAs to inhibit their activity [180], representing another layer of post-transcriptional regulation that CSCs can exploit to promote tumor progression. LUAD CSC-like cells produce exosomes enriched in the lncRNA Mir100hg, which can promote metastasis by enhancing ALDOA activity and N3K14 lactylation [181].
5.2 CSC exosome-mediated immune modulation
Recipients of CSC and non-CSC-derived exosomes are not only limited to cancer cells. CSC-derived exosomes have been demonstrated to mediate immune cell activity and promote immunosuppressive TME. While Wang et al. demonstrate that exosomal miR-210-3p can inhibit FGFRL1 in cancer cells [178], CSC-derived exosomes containing miR-210 also target macrophages in prostate cancer. Inhibition of FGFRL1 in macrophages promotes M2 polarization, while co-injection of these M2-polarized macrophages with prostate cancer cells promotes tumor growth and gemcitabine resistance in vivo [182]. In oral squamous cell carcinoma, CSCs produce exosomes containing the lncRNA UCA1, which inhibits miRNA-134-mediated silencing of laminin γ2 (LAMC2) in macrophages. The resulting upregulation of LAMC2 in macrophages and CD4+ T cells upon exosomal delivery of UCA1 promotes M2-polarization and reduces the proportion of activated IFNγ+/CD4+ T cells [183]. Similarly, tenascin-C, an ECM glycoprotein, is packaged into exosomes by glioblastoma stem cell-like cells to reduce T cell proliferation and activation via tenascin-C/α5β1 and αvβ6-mediated signaling [184].
Furthermore, Gabrusiewicz et al. suggest that only monocytes and activated T cells are able to uptake glioblastoma stem cell-derived exosomes. Recipient monocytes exhibit M2-polarization and increased PD-L1 expression [185], suggesting that CSC-derived exosomes can selectively target certain immune cells within the TME. In CRC, CSC-derived exosomes localize to mouse bone marrow and induce bone marrow-derived neutrophils to secrete IL-1β. These IL-1β-secreting neutrophils exhibit increased lifespan and ability to enhance tumorigenicity of CRC cells in vivo [186]. Similarly, CRC CSCs produce exosomes enriched with β-catenin, IL-6, TGF-β1, and pSTAT3, which induce M2 macrophage polarization and normal fibroblast to CAF conversion, and reduction of this exosomal cargo upon ovatodiolide treatment reduces tumor growth in vivo [187]. In vitro, monocytes that differentiate into dendritic cells in the presence of RCC CSC-derived exosomes containing the human leukocyte antigen G (HLA-G) exhibit lower levels of dendritic cell activation markers CD83 and CD40, and the MHC-II molecule, HLA-DR [188].
5.3 CSC exosome-mediated chemoresistance
CSC-derived exosomes have also been suggested to mediate chemoresistance. Oral squamous cell carcinoma exosomes promote resistance to cisplatin in vitro, while reduction of exosomal cargo via ovatodiolide treatment sensitizes tumors to cisplatin in vivo [189]. Exosomes derived from gemcitabine-resistant pancreatic CSCs promote gemcitabine resistance in gemcitabine-sensitive cells by transferring miR-210 [190]. Similarly, in hepatocellular carcinoma, CSC-derived exosomes enhance regorafenib resistance in vitro in a Nanog-dependent manner [191], suggesting that CSC-derived exosomes may be another mechanism through which tumors can develop chemoresistance. Additionally, more substantial evidence demonstrates dysregulation of exosome production upon chemotherapy treatment. Breast CSC-like cells treated with cisplatin differentially express numerous miRNAs whose miRNA transcripts include numerous cancer-relevant proteins such as MYC and JAG1 [192]. Likewise, docetaxel and doxorubicin treatment of non-CSC breast cancer cells induces production of exosomal miRNAs targeting the transcription factor ONECUT2. While in vivo data suggests that ONECUT2 may promote tumor growth, miRNA-mediated downregulation of ONECUT2 upon docetaxel/doxorubicin treatment induces a stem-like phenotype by upregulating Notch1, Oct4, and SOX9 in breast cancer cells [193]. In gastric cancer cells, cisplatin and paclitaxel treatment induces exosomal lncFERO secretion, which promotes SCD1 mRNA translation to protect recipient CSCs against ferroptosis [194]. In bladder cancer, treatment of non-CSCs with gemcitabine/cisplatin promotes the production of exosomes that can protect recipient CSCs from chemotherapy [195].
5.4 Non-cancer cell-derived exosomes
Within the TME, the production of exosomes is not limited to cancer cells alone. In colorectal cancer, CAFs secrete exosomal lncRNA H19, which acts as a miR-141 sponge to promote tumor growth and resistance to oxaliplatin in recipient cells [196]. These non-cancer cell-derived exosomes have also been demonstrated to mediate stemness. In CRC, CAF-derived exosomes containing Wnt3a enrich stemness markers in cancer cells, promote their sphere formation ability, and drive in-vivo chemoresistance and tumor proliferation [197], while MDSC-derived exosomes containing S100A9 promote stemness of CRC cells in vitro and tumor growth in vivo [198]. Wang et al. demonstrate that S100A9 binds NADPH oxidase to increase ROS production and subsequently activate STAT3 and NF-κB signaling. Subsequent work by Wang et al. demonstrates that MDSC-derived exosomes also promote M2-macrophage polarization by transferring miR-93-5p to downregulate STAT3 signaling in recipient monocytes [199]. In addition to CAFs and MDSCs, BM-MSCs also regulate CSCs via exosomal signaling. BM-MSCs exosomes containing miR-142-3p promote sphere formation, stemness marker expression, and the proportion of CD133+/LGR5+ CSCs in vitro, while CRC cells overexpressing miR-142-3p exhibit increased tumorigenesis in vivo. Mechanistically, miR-142-3p targets mRNA transcripts of DUMB, a negative regulator of Notch signaling, to promote Notch pathway activity [200]. Similarly, in acute myeloid leukemia, BM-MSCs-derived exosomes upregulate S100A4 expression in recipient cancer cells, enhancing their sphere formation ability and expression of stemness markers [201]. However, BM-MSC exosomes containing the circular RNA circ-0030167 inhibit stemness in vitro, while also reducing tumor growth in vivo by inhibiting miR-338-5p/WIF1/Wnt8/β-catenin signaling in pancreatic cancer cells [202]. Altogether, these findings suggest that apart from CSCs and non-CSCs, various stromal and immune cells also contribute to the exosomal secretome within the TME.
6. Endothelial Cells
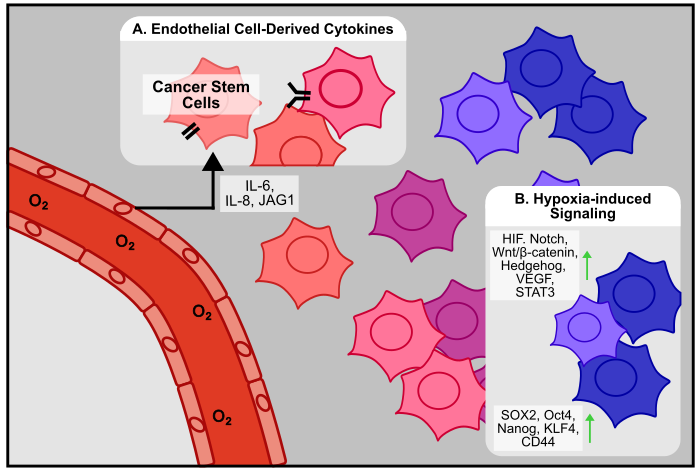
Emerging evidence indicates that endothelial cells, beyond forming new vasculature, also actively contribute to CSC maintenance through direct and paracrine interactions (Figure 6A). In brain tumors, Nestin+/CD34- CSCs are associated with endothelial cells in vivo, and also physically interact in vitro. Co-culture experiments demonstrated that endothelial cells enhance CSC self-renewal, while co-injection of CSCs and endothelial cells in vivo promotes tumor proliferation [203]. A similar phenomenon has been demonstrated in CRC, where endothelial cells are associated with CD133+ CSCs in vivo, enrich the proportion of ALDH1+ and CD133+ CSCs, and dedifferentiate cancer cells to CSCs via secretion of Notch ligand Jagged1 in vitro [204]. In GBM, endothelial cell-derived IL-8 is responsible for promoting stemness and tumor growth in vivo [205]. Similarly, in HNSCC, endothelial cells enhance cancer cell stemness via secretion of IL-6, and co-injection of endothelial cells with ALDH+/CD44+ CSCs increases tumorigenesis in vivo [206]. While ample evidence demonstrates the close association and interaction between endothelial cells and CSCs, Conigliaro et al. show that in HCC, CSC-derived exosomes containing the long noncoding RNA lncRNA H19 are responsible for promoting the attachment of CSCs to endothelial cells [207].
7. Hypoxia
Within the human body, oxygen is required for most biological processes. As such, cells have evolved mechanisms to sense and respond to low concentrations of oxygen, also known as hypoxia. The central hypoxia pathway involves the hypoxia inducible factor (HIF) family of transcription factors. The canonic HIF complex is composed of an HIF-α and an HIF-β subunit, which form a heterodimeric transcription factor to activate hypoxia-response translation [208]. In the most widely studied HIF complex, HIF-1, the β subunit HIF-1β is stably expressed while the α subunit HIF-1α exhibits oxygen-dependent regulation [209]. Under normal concentrations of oxygen, or normoxia, HIF-1α is hydroxylated at key residues, which allows a component of the E3 ubiquitin ligase, the von Hippel-Lindau (VHL) protein, to bind and target HIF-1α for proteasomal degradation. Under hypoxia, HIF-1α hydroxylation is inhibited, and the transcription factor is stabilized and free to complex with HIF-1β to regulate gene expression [210]. HIF regulates numerous cellular functions involving metabolism, gene transcription and translation, and angiogenesis [211]. However, given that hypoxia is a frequent characteristic of solid tumors, HIF signaling is, unsurprisingly, aberrantly activated within the tumor microenvironment to regulate many aspects of cancer biology, including cancer stem cell maintenance (Figure 6B) [212,213]. Within the tumor microenvironment, fast-growing cancer cells can outpace oxygen supply, leading to the development of hypoxic regions [214]. Given that hypoxia has been shown to be an important regulator of embryonic development by promoting dedifferentiation to a stem-like state [215], it is unsurprising that hypoxia has been shown to similarly regulate cancer stemness in the TME (Figure 6B). In glioblastoma, hepatocellular carcinoma, and lung cancer, hypoxia promotes expression of stemness markers in vitro. In sorted CD133- non-CSC cells, long-term hypoxia treatment enriches the population of CD133+ CSC cells [216], suggesting that hypoxia may be a source of new stem-like cells within the TME. These findings have been reaffirmed in ovarian cancer, where hypoxia-induced NOTCH signaling enhances SOX2 expression, chemoresistance, and sphere formation [217]. Similarly, in GBM, hypoxia promotes the proliferation and maintenance of GSCs via an HIF-1α-dependent stabilization of the NOTCH ligand NICD [218]. Hypoxia has also been shown to regulate the stem-cell relevant Wnt/β-catenin pathway. In colorectal cancer, hypoxia activates Wnt/β-catenin signaling to promote sphere formation and the expression of stemness markers in vitro [219]. However, in GBM, hypoxia-dependent inhibition of the β-catenin/TCF-4 complex allows for the formation of the competing HIF-1α/TCF1/β-catenin complex to promote neuronal differentiation away from a stem-like state [220], suggesting that the numerous binding partners of HIF-1α and β-catenin complexes may act to fine-tune downstream gene expression in a tissue-dependent manner.
Hypoxia has also been shown to induce Hh signaling via HIF-1α [221]. In myeloid leukemia, CSCs rely on Hh signaling to maintain stemness [222]. Likewise, inhibition of Hh signaling in CD133+ GSCs decreases stemness markers and secondary sphere formation ability, suggesting that hypoxia-dependent Hh signaling may also play a role in GSC maintenance [223]. In pancreatic cancer, hypoxia can induce EMT through a mechanism that is Hh ligand independent, but dependent on HIF-1α and the Hh pathway protein Smoothened (SMO) [224]. Interestingly, hypoxia causes cancer cells to secrete the Hh ligand Sonic hedgehog (Shh), which acts on surrounding fibroblasts to synthesize fibronectin and collagen [225], suggesting that hypoxia may also play a role in mediating cancer cell-NF/CAF communication to further promote stemness within the TME. Apart from HIF-1α, HIF-2α has also been shown to contribute to CSC maintenance. In CRC, chronic hypoxia induces higher expression of HIF-2α, resulting in higher expression of stemness genes KLF4 and Oct4 [226]. In GBM, HIF-1α and HIF-2α both contribute to glioma cell dedifferentiation, while HIF-2α knockout decreases SOX2 expression and the proportion of CD133+/CD15+ CSCs [227]. Similarly, breast CSCs have also been shown to rely on HIF-2α for self-renewal and sphere formation ability, and HIF-2α knockdown in vivo synergizes with paclitaxel to suppress tumor growth [228]. While numerous stemness genes like SOX2 and Oct4 are likely regulated far downstream of HIF-1α, vascular endothelial growth factor (VEGF) contains a hypoxia response element (HRE) consensus sequence in its promoter and is known to be directly regulated by the HIF-1 transcription factor [229]. In triple negative breast cancer, VEGF promotes mammosphere formation, increases the proportion of ALDH+ CSCs, and upregulates SOX2 expression in a STAT3 and Myc-dependent manner [230]. Likewise, breast CSCs express higher levels of the VEGF receptor, Neuropilin-2 (NRP2) [231], further suggesting that the HIF-1α/VEGF/NRP2 axis contributes to stemness. Unsurprisingly, inhibition of NRP2 in breast CSCs inhibits self-renewal and mammosphere formation ability [232].
8. Conclusions
Within the context of the TME, CSCs and stemness appear to be highly dynamic. While CSCs may possess intrinsically higher activity in pathways that promote self-renewal and stemness, a multitude of signals from the TME also plays a significant role in maintaining stemness. Cytokines, growth factors, and other exosome-derived molecules such as miRNAs secreted by CAFs; MSCs; immune cells such as MDSCs, TAMs, and T cells; and other non-CSCs and CSCs contribute to the activation of numerous stemness-promoting pathways in CSCs. In turn, CSCs themselves contribute to the TME signaling landscape to recruit and polarize TAMs, convert normal fibroblasts to CAFs, promote angiogenesis, and suppress immune activity. Clearly, regulation of CSCs and stemness is a complex and reciprocal process.
While CSCs may originate during tumorigenesis from transformation of tissue-resident stem cells, dedifferentiation of epithelial or stromal cells, or other events such as cell fusion and EMT [233], the emergence of new CSCs and regions enriched in high-stemness cells may be driven by environmental cues from the TME. Similar to normal human stem cells, which exist in niches that support and regulate their ability to self-renew and differentiate, CSCs appear to depend on analogous supportive niches within the TME. For example, regulation of normal intestinal stem cells relies on the intestinal crypt, which provides a niche for stem cell development. Cells at the base of the intestinal crypt are exposed to high levels of Wnt, Notch, and EGF ligands, which contribute to the maintenance of stem-like properties [234]. Likewise, in the TME, regions of hypoxia may provide a niche for the development of stem-like cancer cells. Activation of HIF signaling and downstream Wnt/β-catenin signaling within cancer cells residing in these hypoxic regions generates cancer cells exhibiting stem-like properties. Within this hypoxic niche, these CSC-like cells may recruit other cells such as endothelial cells, which can further contribute to promoting stemness.
Like the diverse array of normal stem cell niches such as neural stem cells within the subventricular zone, hair follicle stem cells in the hair follicle bulge, and hematopoietic stem cells in the bone marrow, numerous environmental and cellular factors contribute to the varied mechanisms underlying CSC niches [234–236]. Notably, CSCs appear to possess the ability to actively create and remodel their own supportive niches within the TME. By recruiting circulating cells such as MDSCs, Tregs, and monocytes, and by reprogramming immune cells, converting normal fibroblasts to CAFs, and polarizing monocytes into tumor-promoting macrophages, CSCs can establish a microenvironment that reinforces their stem-like state. Within these constructed niches, these recruited/transformed cells are not only able to help maintain stemness, but also suppress immune activity, promote metastasis, contribute to treatment resistance, and overall contribute to tumor progression. Given this, targeting cancer stem cells appears to be a promising avenue for cancer therapy, and countless therapeutic strategies have been developed to directly target cancer stem cells. Two general approaches involve inhibition of CSC-related pathways such as Wnt, Notch, and Hh pathways, and immunotherapy-based strategies against CSC markers such as CD44, EpCAM, and CD33 [237,238]. While some have shown some promise in clinic, no CSC-based therapies have significantly transformed standard of care and outcomes [237]. As such, the crosstalk between CSCs and TME provides countless new targets for cancer therapy. Instead of directly targeting CSCs, targeting CAFs, TAMs, etc. within the TME can disrupt the niche that helps promote cancer stemness. Where directly targeting CSC-related pathways may be infeasible due to poor selectivity, undruggable targets, etc., targeting TME components that produce activators of such pathways can provide additional flexibility. Similarly, when CSC markers may be non-specific and easily lost, resulting in therapy resistance, targeting cellular markers within the TME could provide another more specific and stable target for reducing CSCs.
Declarations
Ethics Statement
Not applicable
Consent for Publication
Not applicable
Availability of Data and material
Not applicable
Funding
This work was supported by grants from the NIH (R01CA248027, R01CA211175, and R01CA234124) and CDMRP (W81XWH211028, and HT9425-24-1-1034)
Competing Interests
The authors have declared that no competing interests exist.
Abbreviations
The following abbreviations are used in this manuscript:
- ALDH:
- Aldehyde dehydrogenase
- CAF:
- Cancer-associated fibroblast
- CRC:
- Colorectal cancer
- CSC:
- Cancer stem cell
- CSF:
- Colony stimulating factor
- ECM:
- Extracellular matrix
- EMT:
- Epithelial-mesenchymal transition
- FGF:
- Fibroblast growth factor
- GBM:
- Glioblastoma
- GSC:
- Glioma stem cell
- HCC:
- Hepatocellular carcinoma
- HGF:
- hepatocyte growth factor
- Hh:
- Hedgehog
- HIF:
- Hypoxia inducible factor
- HNSCC:
- Head and neck squamous cell carcinoma
- IHC:
- Immunohistochemistry
- LUAD:
- Lung adenocarcinoma
- MDSC:
- Myeloid-derived suppressive cell
- M-MDSC:
- Monocytic MDSC
- MMP:
- Matrix metalloproteinase
- MSC:
- Mesenchymal stem cell
- NK:
- Natural killer
- NO:
- Nitric oxide
- NSCLC:
- Non-small cell lung cancer
- OSCC:
- Oral squamous cell carcinoma
- PDAC:
- Pancreatic Ductal Adenocarcinoma
- PGE2:
- Prostaglandin E2
- PMN-MDSC:
- Polymorphonuclear MDSC
- RCC:
- Renal cell carcinoma
- Shh:
- Sonic hedgehog
- TAM:
- Tumor associated macrophages
- TME:
- Tumor microenvironment
- TNFα:
- Tumor necrosis factor α
- Treg:
- Regulatory T cell
References
| 1. |
Atashzar MR, Baharlou R, Karami J, Abdollahi H, Rezaei R, Pourramezan F, et al. Cancer stem cells: A review from origin to therapeutic implications. J Cell Physiol. 2020;235:790-803.
[Google Scholar]
[CrossRef]
|
| 2. |
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-648.
[Google Scholar]
[CrossRef]
|
| 3. |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-3988.
[Google Scholar]
[CrossRef]
|
| 4. |
Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267-284.
[Google Scholar]
[CrossRef]
|
| 5. |
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018;2018:5416923.
[Google Scholar]
[CrossRef]
|
| 6. |
Shiozawa Y, Nie B, Pienta KJ, Morgan TM, Taichman RS. Cancer stem cells and their role in metastasis. Pharmacol Ther. 2013;138:285-293.
[Google Scholar]
[CrossRef]
|
| 7. |
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
[Google Scholar]
[CrossRef]
|
| 8. |
Prager BC, Xie Q, Bao S, Rich JN. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell. 2019;24:41-53.
[Google Scholar]
[CrossRef]
|
| 9. |
van Neerven SM, Tieken M, Vermeulen L, Bijlsma MF. Bidirectional interconversion of stem and non-stem cancer cell populations: A reassessment of theoretical models for tumor heterogeneity. Mol Cell Oncol. 2016;3:e1098791.
[Google Scholar]
[CrossRef]
|
| 10. |
S SF, Szczesna K, Iliou MS, Al-Qahtani M, Mobasheri A, Kobolak J, et al. In vitro models of cancer stem cells and clinical applications. BMC Cancer. 2016;16:738.
[Google Scholar]
[CrossRef]
|
| 11. |
Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. 2024;31:617-639.
[Google Scholar]
[CrossRef]
|
| 12. |
de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374-403.
[Google Scholar]
[CrossRef]
|
| 13. |
Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126-133.
[Google Scholar]
[CrossRef]
|
| 14. |
Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5:166.
[Google Scholar]
[CrossRef]
|
| 15. |
Bejarano L, Jordao MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11:933-959.
[Google Scholar]
[CrossRef]
|
| 16. |
Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 2021;184:3852-3872.
[Google Scholar]
[CrossRef]
|
| 17. |
Lendahl U, Muhl L, Betsholtz C. Identification, discrimination and heterogeneity of fibroblasts. Nat Commun. 2022;13:3409.
[Google Scholar]
[CrossRef]
|
| 18. |
Deng CC, Hu YF, Zhu DH, Cheng Q, Gu JJ, Feng QL, et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat Commun. 2021;12:3709.
[Google Scholar]
[CrossRef]
|
| 19. |
Luo H, Xia X, Huang LB, An H, Cao M, Kim GD, et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun. 2022;13:6619.
[Google Scholar]
[CrossRef]
|
| 20. |
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174-186.
[Google Scholar]
[CrossRef]
|
| 21. |
Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther. 2021;6:218.
[Google Scholar]
[CrossRef]
|
| 22. |
Miyazaki Y, Oda T, Inagaki Y, Kushige H, Saito Y, Mori N, et al. Adipose-derived mesenchymal stem cells differentiate into heterogeneous cancer-associated fibroblasts in a stroma-rich xenograft model. Sci Rep. 2021;11:4690.
[Google Scholar]
[CrossRef]
|
| 23. |
Kular JK, Basu S, Sharma RI. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J Tissue Eng. 2014;5:2041731414557112.
[Google Scholar]
[CrossRef]
|
| 24. |
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120.
[Google Scholar]
[CrossRef]
|
| 25. |
Cavaco ACM, Rezaei M, Caliandro MF, Lima AM, Stehling M, Dhayat SA, et al. The Interaction between Laminin-332 and alpha3beta1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers (Basel). 2018;11.
[Google Scholar]
[CrossRef]
|
| 26. |
Govaere O, Wouters J, Petz M, Vandewynckel YP, Van den Eynde K, Van den Broeck A, et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J Hepatol. 2016;64:609-617.
[Google Scholar]
[CrossRef]
|
| 27. |
Hao N, Yang D, Liu T, Liu S, Lu X, Chen L. Laminin-integrin a6b4 interaction activates notch signaling to facilitate bladder cancer development. BMC Cancer. 2022;22:558.
[Google Scholar]
[CrossRef]
|
| 28. |
Qi X, Zhou J, Wang P, Li Y, Li H, Miao Y, et al. KLF7-regulated ITGA2 as a therapeutic target for inhibiting oral cancer stem cells. Cell Death Dis. 2025;16:354.
[Google Scholar]
[CrossRef]
|
| 29. |
Zhong C, Tao B, Tang F, Yang X, Peng T, You J, et al. Remodeling cancer stemness by collagen/fibronectin via the AKT and CDC42 signaling pathway crosstalk in glioma. Theranostics. 2021;11:1991-2005.
[Google Scholar]
[CrossRef]
|
| 30. |
Wu JL, Xu CF, Yang XH, Wang MS. Fibronectin promotes tumor progression through integrin alphavbeta3/PI3K/AKT/SOX2 signaling in non-small cell lung cancer. Heliyon. 2023;9:e20185.
[Google Scholar]
[CrossRef]
|
| 31. |
Zhou F, Sun J, Ye L, Jiang T, Li W, Su C, et al. Fibronectin promotes tumor angiogenesis and progression of non-small-cell lung cancer by elevating WISP3 expression via FAK/MAPK/ HIF-1alpha axis and activating wnt signaling pathway. Exp Hematol Oncol. 2023;12:61.
[Google Scholar]
[CrossRef]
|
| 32. |
Park J, Schwarzbauer JE. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene. 2014;33:1649-1657.
[Google Scholar]
[CrossRef]
|
| 33. |
Li CL, Yang D, Cao X, Wang F, Hong DY, Wang J, et al. Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain. Oncol Lett. 2017;13:3889-3895.
[Google Scholar]
[CrossRef]
|
| 34. |
Wang X, Zhou Y, Wang Y, Yang J, Li Z, Liu F, et al. Overcoming cancer treatment resistance: Unraveling the role of cancer-associated fibroblasts. J Natl Cancer Cent. 2025;5(3):237-251.
[Google Scholar]
[CrossRef]
|
| 35. |
Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:201-212.
[Google Scholar]
[CrossRef]
|
| 36. |
Yu WH, Wu E, Li Y, Hou HH, Yu SC, Huang PT, et al. Matrix Metalloprotease-7 Mediates Nucleolar Assembly and Intra-nucleolar Cleaving p53 in Gefitinib-Resistant Cancer Stem Cells. iScience. 2020;23:101600.
[Google Scholar]
[CrossRef]
|
| 37. |
Sapudom J, Muller CD, Nguyen KT, Martin S, Anderegg U, Pompe T. Matrix Remodeling and Hyaluronan Production by Myofibroblasts and Cancer-Associated Fibroblasts in 3D Collagen Matrices. Gels. 2020;6(4):33.
[Google Scholar]
[CrossRef]
|
| 38. |
Ouhtit A, Rizeq B, Saleh HA, Rahman MM, Zayed H. Novel CD44-downstream signaling pathways mediating breast tumor invasion. Int J Biol Sci. 2018;14:1782-1790.
[Google Scholar]
[CrossRef]
|
| 39. |
Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11:64.
[Google Scholar]
[CrossRef]
|
| 40. |
Gomez KE, Wu F, Keysar SB, Morton JJ, Miller B, Chimed TS, et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020;80:4185-4198.
[Google Scholar]
[CrossRef]
|
| 41. |
Safaei S, Sajed R, Shariftabrizi A, Dorafshan S, Saeednejad Zanjani L, Dehghan Manshadi M, et al. Tumor matrix stiffness provides fertile soil for cancer stem cells. Cancer Cell Int. 2023;23:143.
[Google Scholar]
[CrossRef]
|
| 42. |
Ma D, Luo Q, Song G. Matrix stiffening facilitates stemness of liver cancer stem cells by YAP activation and BMF inhibition. Biomater Adv. 2024;163:213936.
[Google Scholar]
[CrossRef]
|
| 43. |
Kim D, You E, Jeong J, Ko P, Kim JW, Rhee S. DDR2 controls the epithelial-mesenchymal-transition-related gene expression via c-Myb acetylation upon matrix stiffening. Sci Rep. 2017;7:6847.
[Google Scholar]
[CrossRef]
|
| 44. |
Tan F, Huang Y, Pei Q, Liu H, Pei H, Zhu H. Matrix stiffness mediates stemness characteristics via activating the Yes-associated protein in colorectal cancer cells. J Cell Biochem. 2019;120:2213-2225.
[Google Scholar]
[CrossRef]
|
| 45. |
Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans. 2017;45:229-236.
[Google Scholar]
[CrossRef]
|
| 46. |
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897.
[Google Scholar]
[CrossRef]
|
| 47. |
Suh J, Kim DH, Lee YH, Jang JH, Surh YJ. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol Carcinog. 2020;59:1028-1040.
[Google Scholar]
[CrossRef]
|
| 48. |
Santolla MF, Vivacqua A, Lappano R, Rigiracciolo DC, Cirillo F, Galli GR, et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells. 2019;8:223.
[Google Scholar]
[CrossRef]
|
| 49. |
Wang Z, Chen C, Sun L, He M, Huang T, Zheng J, et al. Fibroblast growth factor 2 promotes osteo/odontogenic differentiation in stem cells from the apical papilla by inhibiting PI3K/AKT pathway. Sci Rep. 2024;14:19354.
[Google Scholar]
[CrossRef]
|
| 50. |
Sheta M, Hassan G, Afify SM, Monzur S, Kumon K, Abu Quora HA, et al. Chronic exposure to FGF2 converts iPSCs into cancer stem cells with an enhanced integrin/focal adhesion/PI3K/AKT axis. Cancer Lett. 2021;521:142-154.
[Google Scholar]
[CrossRef]
|
| 51. |
Duan JJ, Qiu W, Xu SL, Wang B, Ye XZ, Ping YF, et al. Strategies for isolating and enriching cancer stem cells: well begun is half done. Stem Cells Dev. 2013;22:2221-2239.
[Google Scholar]
[CrossRef]
|
| 52. |
Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. 2014;25:15-22.
[Google Scholar]
[CrossRef]
|
| 53. |
Wang B, Liu W, Liu C, Du K, Guo Z, Zhang G, et al. Cancer-Associated Fibroblasts Promote Radioresistance of Breast Cancer Cells via the HGF/c-Met Signaling Pathway. Int J Radiat Oncol Biol Phys. 2023;116:640-654.
[Google Scholar]
[CrossRef]
|
| 54. |
Ding X, Ji J, Jiang J, Cai Q, Wang C, Shi M, et al. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018;9:867.
[Google Scholar]
[CrossRef]
|
| 55. |
Zhang R, Qi F, Shao S, Li G, Feng Y. Human colorectal cancer-derived carcinoma associated fibroblasts promote CD44-mediated adhesion of colorectal cancer cells to endothelial cells by secretion of HGF. Cancer Cell Int. 2019;19:192.
[Google Scholar]
[CrossRef]
|
| 56. |
Loh JJ, Li TW, Zhou L, Wong TL, Liu X, Ma VWS, et al. FSTL1 Secreted by Activated Fibroblasts Promotes Hepatocellular Carcinoma Metastasis and Stemness. Cancer Res. 2021;81:5692-5705.
[Google Scholar]
[CrossRef]
|
| 57. |
Cheng S, Huang Y, Lou C, He Y, Zhang Y, Zhang Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol Ther. 2019;20:328-337.
[Google Scholar]
[CrossRef]
|
| 58. |
Wu M, Ding Y, Wu N, Jiang J, Huang Y, Zhang F, et al. FSTL1 promotes growth and metastasis in gastric cancer by activating AKT related pathway and predicts poor survival. Am J Cancer Res. 2021;11:712-728.
[Google Scholar]
|
| 59. |
Jing SY, Liu D, Feng N, Dong H, Wang HQ, Yan X, et al. Spatial multiomics reveals a subpopulation of fibroblasts associated with cancer stemness in human hepatocellular carcinoma. Genome Med. 2024;16:98.
[Google Scholar]
[CrossRef]
|
| 60. |
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017;8:20741-20750.
[Google Scholar]
[CrossRef]
|
| 61. |
Wang CQ, Sun HT, Gao XM, Ren N, Sheng YY, Wang Z, et al. Interleukin-6 enhances cancer stemness and promotes metastasis of hepatocellular carcinoma via up-regulating osteopontin expression. Am J Cancer Res. 2016;6:1873-1889.
[Google Scholar]
|
| 62. |
Guo Z, Zhang H, Fu Y, Kuang J, Zhao B, Zhang L, et al. Cancer-associated fibroblasts induce growth and radioresistance of breast cancer cells through paracrine IL-6. Cell Death Discov. 2023;9:6.
[Google Scholar]
[CrossRef]
|
| 63. |
Cheteh EH, Sarne V, Ceder S, Bianchi J, Augsten M, Rundqvist H, et al. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020;6:42.
[Google Scholar]
[CrossRef]
|
| 64. |
Zhai J, Shen J, Xie G, Wu J, He M, Gao L, et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019;454:37-43.
[Google Scholar]
[CrossRef]
|
| 65. |
Gu X, Zhu Y, Su J, Wang S, Su X, Ding X, et al. Lactate-induced activation of tumor-associated fibroblasts and IL-8-mediated macrophage recruitment promote lung cancer progression. Redox Biol. 2024;74:103209.
[Google Scholar]
[CrossRef]
|
| 66. |
Ji Z, Tian W, Gao W, Zang R, Wang H, Yang G. Cancer-Associated Fibroblast-Derived Interleukin-8 Promotes Ovarian Cancer Cell Stemness and Malignancy Through the Notch3-Mediated Signaling. Front Cell Dev Biol. 2021;9:684505.
[Google Scholar]
[CrossRef]
|
| 67. |
Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. 2019;378:131-138.
[Google Scholar]
[CrossRef]
|
| 68. |
Ma Z, Yu D, Tan S, Li H, Zhou F, Qiu L, et al. CXCL12 alone is enough to Reprogram Normal Fibroblasts into Cancer-Associated Fibroblasts. Cell Death Discov. 2025;11:156.
[Google Scholar]
[CrossRef]
|
| 69. |
Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103:1671-1679.
[Google Scholar]
[CrossRef]
|
| 70. |
Zielinska KA, Katanaev VL. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers (Basel). 2020;12:3071.
[Google Scholar]
[CrossRef]
|
| 71. |
Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72:2768-2779.
[Google Scholar]
[CrossRef]
|
| 72. |
Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45-53.
[Google Scholar]
[CrossRef]
|
| 73. |
Ostroumov D, Fekete-Drimusz N, Saborowski M, Kuhnel F, Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci. 2018;75:689-713.
[Google Scholar]
[CrossRef]
|
| 74. |
Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci. 2021;22:6995.
[Google Scholar]
[CrossRef]
|
| 75. |
Chen P, Hsu WH, Han J, Xia Y, DePinho RA. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021;34:108597.
[Google Scholar]
[CrossRef]
|
| 76. |
Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:127.
[Google Scholar]
[CrossRef]
|
| 77. |
Chaintreuil P, Kerreneur E, Bourgoin M, Savy C, Favreau C, Robert G, et al. The generation, activation, and polarization of monocyte-derived macrophages in human malignancies. Front Immunol. 2023;14:1178337.
[Google Scholar]
[CrossRef]
|
| 78. |
Wang S, Wang J, Chen Z, Luo J, Guo W, Sun L, et al. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ precision oncology. 2024;8:31.
[Google Scholar]
[CrossRef]
|
| 79. |
Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res. 2016;119:414-417.
[Google Scholar]
[CrossRef]
|
| 80. |
Zeng J, Liu Z, Sun S, Xie J, Cao L, Lv P, et al. Tumor-associated macrophages recruited by periostin in intrahepatic cholangiocarcinoma stem cells. Oncol Lett. 2018;15:8681-8686.
[Google Scholar]
[CrossRef]
|
| 81. |
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-182.
[Google Scholar]
[CrossRef]
|
| 82. |
Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113-1125.
[Google Scholar]
[CrossRef]
|
| 83. |
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells through a Complex Chemokine Network. Cancer Res. 2019;79:3360-3371.
[Google Scholar]
[CrossRef]
|
| 84. |
Chen W, Chen M, Hong L, Xiahenazi A, Huang M, Tang N, et al. M2-like tumor-associated macrophage-secreted CCL2 facilitates gallbladder cancer stemness and metastasis. Exp Hematol Oncol. 2024;13:83.
[Google Scholar]
[CrossRef]
|
| 85. |
Raghavan S, Mehta P, Xie Y, Lei YL, Mehta G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J Immunother Cancer. 2019;7:190.
[Google Scholar]
[CrossRef]
|
| 86. |
Yao Y, Ye H, Qi Z, Mo L, Yue Q, Baral A, et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin Cancer Res. 2016;22:2778-2790.
[Google Scholar]
[CrossRef]
|
| 87. |
Tao W, Chu C, Zhou W, Huang Z, Zhai K, Fang X, et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat Commun. 2020;11:3015.
[Google Scholar]
[CrossRef]
|
| 88. |
Zhou F, Tao J, Gou H, Liu S, Yu D, Zhang J, et al. FSTL1 sustains glioma stem cell stemness and promotes immunosuppressive macrophage polarization in glioblastoma. Cancer Lett. 2024;611:217400.
[Google Scholar]
[CrossRef]
|
| 89. |
Zhang X, Chen L, Dang WQ, Cao MF, Xiao JF, Lv SQ, et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab Invest. 2020;100:619-629.
[Google Scholar]
[CrossRef]
|
| 90. |
Chen X, Yang M, Yin J, Li P, Zeng S, Zheng G, et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/beta-catenin signaling. Cell Commun Signal. 2022;20:92.
[Google Scholar]
[CrossRef]
|
| 91. |
Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. 2020;11:234.
[Google Scholar]
[CrossRef]
|
| 92. |
Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393-1404.
[Google Scholar]
[CrossRef]
|
| 93. |
Matsubara E, Yano H, Pan C, Komohara Y, Fujiwara Y, Zhao S, et al. The Significance of SPP1 in Lung Cancers and Its Impact as a Marker for Protumor Tumor-Associated Macrophages. Cancers (Basel). 2023;15:2250.
[Google Scholar]
[CrossRef]
|
| 94. |
Fan G, Xie T, Li L, Tang L, Han X, Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. 2024;8:75.
[Google Scholar]
[CrossRef]
|
| 95. |
Yang Y, Wu T, Wang Y, Luo D, Zhao Z, Sun H, et al. Hypoxic tumour-derived exosomal miR-1290 exacerbates the suppression of CD8+ T cells by promoting M2 macrophage polarization. Immunology. 2024;173:672-688.
[Google Scholar]
[CrossRef]
|
| 96. |
Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun. 2022;13:1742.
[Google Scholar]
[CrossRef]
|
| 97. |
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12:86.
[Google Scholar]
[CrossRef]
|
| 98. |
Pu J, Xu Z, Nian J, Fang Q, Yang M, Huang Y, et al. M2 macrophage-derived extracellular vesicles facilitate CD8+T cell exhaustion in hepatocellular carcinoma via the miR-21-5p/YOD1/YAP/beta-catenin pathway. Cell Death Discov. 2021;7:182.
[Google Scholar]
[CrossRef]
|
| 99. |
Honkanen TJ, Tikkanen A, Karihtala P, Makinen M, Vayrynen JP, Koivunen JP. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci Rep. 2019;9:10961.
[Google Scholar]
[CrossRef]
|
| 100. |
Oshi M, Tokumaru Y, Asaoka M, Yan L, Satyananda V, Matsuyama R, et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci Rep. 2020;10:16554.
[Google Scholar]
[CrossRef]
|
| 101. |
Guo L, Cheng X, Chen H, Chen C, Xie S, Zhao M, et al. Induction of breast cancer stem cells by M1 macrophages through Lin-28B-let-7-HMGA2 axis. Cancer Lett. 2019;452:213-225.
[Google Scholar]
[CrossRef]
|
| 102. |
Kainulainen K, Niskanen EA, Kinnunen J, Maki-Mantila K, Hartikainen K, Paakinaho V, et al. Secreted factors from M1 macrophages drive prostate cancer stem cell plasticity by upregulating NANOG, SOX2, and CD44 through NFkappaB-signaling. Oncoimmunology. 2024;13:2393442.
[Google Scholar]
[CrossRef]
|
| 103. |
Shang S, Yang C, Chen F, Xiang RS, Zhang H, Dai SY, et al. ID1 expressing macrophages support cancer cell stemness and limit CD8(+) T cell infiltration in colorectal cancer. Nat Commun. 2023;14:7661.
[Google Scholar]
[CrossRef]
|
| 104. |
Wang Q, Qin Y, Li B. CD8(+) T cell exhaustion and cancer immunotherapy. Cancer Lett. 2023;559:216043.
[Google Scholar]
[CrossRef]
|
| 105. |
Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:e1792.
[Google Scholar]
[CrossRef]
|
| 106. |
Zhang C, Wang H, Wang X, Zhao C, Wang H. CD44, a marker of cancer stem cells, is positively correlated with PD-L1 expression and immune cells infiltration in lung adenocarcinoma. Cancer Cell Int. 2020;20:583.
[Google Scholar]
[CrossRef]
|
| 107. |
Min HY, Cho J, Sim JY, Boo HJ, Lee JS, Lee SB, et al. S100A14: A novel negative regulator of cancer stemness and immune evasion by inhibiting STAT3-mediated programmed death-ligand 1 expression in colorectal cancer. Clin Transl Med. 2022;12:e986.
[Google Scholar]
[CrossRef]
|
| 108. |
Mansour FA, Al-Mazrou A, Al-Mohanna F, Al-Alwan M, Ghebeh H. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology. 2020;9:1729299.
[Google Scholar]
[CrossRef]
|
| 109. |
López Flores M, Honrado Franco E, Sánchez Cousido LF, Minguito-Carazo C, Sanz Guadarrama O, López González L, et al. Relationship between Aldehyde Dehydrogenase, PD-L1 and Tumor-Infiltrating Lymphocytes with Pathologic Response and Survival in Breast Cancer. Cancers (Basel). 2022;14:4418.
[Google Scholar]
[CrossRef]
|
| 110. |
Koh YW, Han JH, Haam S. Expression of PD-L1, cancer stem cell and epithelial-mesenchymal transition phenotype in non-small cell lung cancer. Pathology. 2021;53:239-246.
[Google Scholar]
[CrossRef]
|
| 111. |
Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908.
[Google Scholar]
[CrossRef]
|
| 112. |
Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology. 2021;10:1997385.
[Google Scholar]
[CrossRef]
|
| 113. |
Wei F, Zhang T, Deng SC, Wei JC, Yang P, Wang Q, et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019;450:1-13.
[Google Scholar]
[CrossRef]
|
| 114. |
Jin H, Kim HJ. NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations. Int J Mol Sci. 2021;22:9639.
[Google Scholar]
[CrossRef]
|
| 115. |
Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J Immunol. 2015;195:4010-4019.
[Google Scholar]
[CrossRef]
|
| 116. |
Mukherjee S, Chakraborty S, Basak U, Pati S, Dutta A, Dutta S, et al. Breast cancer stem cells generate immune-suppressive T regulatory cells by secreting TGFbeta to evade immune-elimination. Discov Oncol. 2023;14:220.
[Google Scholar]
[CrossRef]
|
| 117. |
Basak U, Chakraborty S, Mukherjee S, Pati S, Khan P, Ghosh S, et al. Breast cancer stem cells convert anti-tumor CD4(+) T cells to pro-tumor T regulatory cells: Potential role of exosomal FOXP3. Cell Immunol. 2025;409–410:104931.
[Google Scholar]
[CrossRef]
|
| 118. |
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9:67-78.
[Google Scholar]
[CrossRef]
|
| 119. |
Chikamatsu K, Takahashi G, Sakakura K, Ferrone S, Masuyama K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck. 2011;33:208-215.
[Google Scholar]
[CrossRef]
|
| 120. |
You Y, Li Y, Li M, Lei M, Wu M, Qu Y, et al. Ovarian cancer stem cells promote tumour immune privilege and invasion via CCL5 and regulatory T cells. Clin Exp Immunol. 2018;191:60-73.
[Google Scholar]
[CrossRef]
|
| 121. |
Xiang T, Long H, He L, Han X, Lin K, Liang Z, et al. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene. 2015;34:165-176.
[Google Scholar]
[CrossRef]
|
| 122. |
Zhang Y, Zoltan M, Riquelme E, Xu H, Sahin I, Castro-Pando S, et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology. 2018;155:210-223 e213.
[Google Scholar]
[CrossRef]
|
| 123. |
Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. 2021;54:875-884.
[Google Scholar]
[CrossRef]
|
| 124. |
Lasser SA, Ozbay Kurt FG, Arkhypov I, Utikal J, Umansky V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. 2024;21:147-164.
[Google Scholar]
[CrossRef]
|
| 125. |
Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6:362.
[Google Scholar]
[CrossRef]
|
| 126. |
Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632-644.
[Google Scholar]
[CrossRef]
|
| 127. |
Shidal C, Singh NP, Nagarkatti P, Nagarkatti M. MicroRNA-92 Expression in CD133(+) Melanoma Stem Cells Regulates Immunosuppression in the Tumor Microenvironment via Integrin-Dependent Activation of TGFbeta. Cancer Res. 2019;79:3622-3635.
[Google Scholar]
[CrossRef]
|
| 128. |
Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6:80-95.
[Google Scholar]
[CrossRef]
|
| 129. |
Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21:184.
[Google Scholar]
[CrossRef]
|
| 130. |
Zhang R, Dong M, Tu J, Li F, Deng Q, Xu J, et al. PMN-MDSCs modulated by CCL20 from cancer cells promoted breast cancer cell stemness through CXCL2-CXCR2 pathway. Signal Transduct Target Ther. 2023;8:97.
[Google Scholar]
[CrossRef]
|
| 131. |
Li X, Wang J, Wu W, Gao H, Liu N, Zhan G, et al. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. FEBS J. 2020;287:5218-5235.
[Google Scholar]
[CrossRef]
|
| 132. |
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76:3156-3165.
[Google Scholar]
[CrossRef]
|
| 133. |
Wang R, Li Y, Tsung A, Huang H, Du Q, Yang M, et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc Natl Acad Sci U S A. 2018;115:E10127-E10136.
[Google Scholar]
[CrossRef]
|
| 134. |
Zhang T, Lei J, Zheng M, Wen Z, Zhou J. Nitric oxide facilitates the S-nitrosylation and deubiquitination of Notch1 protein to maintain cancer stem cells in human NSCLC. J Cell Mol Med. 2024;28:e70203.
[Google Scholar]
[CrossRef]
|
| 135. |
Tuy K, Rickenbacker L, Hjelmeland AB. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. 2021;44:101953.
[Google Scholar]
[CrossRef]
|
| 136. |
Kawano M, Mabuchi S, Matsumoto Y, Sasano T, Takahashi R, Kuroda H, et al. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci Rep. 2015;5:18217.
[Google Scholar]
[CrossRef]
|
| 137. |
Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. 2020;69:2477-2499.
[Google Scholar]
[CrossRef]
|
| 138. |
Ducarouge B, Redavid AR, Victoor C, Chira R, Fonseca A, Hervieu M, et al. Netrin-1 blockade inhibits tumor associated Myeloid-derived suppressor cells, cancer stemness and alleviates resistance to chemotherapy and immune checkpoint inhibitor. Cell Death Differ. 2023;30:2201-2212.
[Google Scholar]
[CrossRef]
|
| 139. |
Liu J, Gao J, Liang Z, Gao C, Niu Q, Wu F, et al. Mesenchymal stem cells and their microenvironment. Stem Cell Res Ther. 2022;13:429.
[Google Scholar]
[CrossRef]
|
| 140. |
Li C, Zhao H, Wang B. Mesenchymal stem/stromal cells: Developmental origin, tumorigenesis and translational cancer therapeutics. Transl Oncol. 2021;14:100948.
[Google Scholar]
[CrossRef]
|
| 141. |
Han Y, Yang J, Fang J, Zhou Y, Candi E, Wang J, et al. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct Target Ther. 2022;7:92.
[Google Scholar]
[CrossRef]
|
| 142. |
Pittenger MF, Discher DE, Peault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22.
[Google Scholar]
[CrossRef]
|
| 143. |
Antoon R, Overdevest N, Saleh AH, Keating A. Mesenchymal stromal cells as cancer promoters. Oncogene. 2024;43:3545-3555.
[Google Scholar]
[CrossRef]
|
| 144. |
Liang W, Chen X, Zhang S, Fang J, Chen M, Xu Y, et al. Mesenchymal stem cells as a double-edged sword in tumor growth: focusing on MSC-derived cytokines. Cell Mol Biol Lett. 2021;26:3.
[Google Scholar]
[CrossRef]
|
| 145. |
McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. 2011;121:3206-3219.
[Google Scholar]
[CrossRef]
|
| 146. |
Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells. 2015;33:2400-2415.
[Google Scholar]
[CrossRef]
|
| 147. |
Mi F, Gong L. Secretion of interleukin-6 by bone marrow mesenchymal stem cells promotes metastasis in hepatocellular carcinoma. Biosci Rep. 2017;37.
[Google Scholar]
[CrossRef]
|
| 148. |
Herzog AE, Warner KA, Zhang Z, Bellile E, Bhagat MA, Castilho RM, et al. The IL-6R and Bmi-1 axis controls self-renewal and chemoresistance of head and neck cancer stem cells. Cell Death Dis. 2021;12:988.
[Google Scholar]
[CrossRef]
|
| 149. |
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47:570-584.
[Google Scholar]
[CrossRef]
|
| 150. |
Wang T, Song P, Zhong T, Wang X, Xiang X, Liu Q, et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene. 2019;38:4932-4947.
[Google Scholar]
[CrossRef]
|
| 151. |
Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71:614-624.
[Google Scholar]
[CrossRef]
|
| 152. |
Wang J, Liu Y, Ding H, Shi X, Ren H. Mesenchymal stem cell-secreted prostaglandin E(2) ameliorates acute liver failure via attenuation of cell death and regulation of macrophage polarization. Stem Cell Res Ther. 2021;12:15.
[Google Scholar]
[CrossRef]
|
| 153. |
Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2:840-855.
[Google Scholar]
[CrossRef]
|
| 154. |
Minematsu H, Afify SM, Sugihara Y, Hassan G, Zahra MH, Seno A, et al. Cancer stem cells induced by chronic stimulation with prostaglandin E2 exhibited constitutively activated PI3K axis. Sci Rep. 2022;12:15628.
[Google Scholar]
[CrossRef]
|
| 155. |
Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene. 2014;33:2768-2778.
[Google Scholar]
[CrossRef]
|
| 156. |
Kong BH, Shin HD, Kim SH, Mok HS, Shim JK, Lee JH, et al. Increased in vivo angiogenic effect of glioma stromal mesenchymal stem-like cells on glioma cancer stem cells from patients with glioblastoma. Int J Oncol. 2013;42:1754-1762.
[Google Scholar]
[CrossRef]
|
| 157. |
Zhu XY, Liu WT, Hou XJ, Zong C, Yu W, Shen ZM, et al. CD34(+)CLDN5(+) tumor associated senescent endothelial cells through IGF2-IGF2R signaling increased cholangiocellular phenotype in hepatocellular carcinoma. J Adv Res. 2024.
[Google Scholar]
[CrossRef]
|
| 158. |
Rubinstein-Achiasaf L, Morein D, Ben-Yaakov H, Liubomirski Y, Meshel T, Elbaz E, et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers (Basel). 2021;13:1472.
[Google Scholar]
[CrossRef]
|
| 159. |
Saxena N, Chakraborty S, Dutta S, Bhardwaj G, Karnik N, Shetty O, et al. Stiffness-dependent MSC homing and differentiation into CAFs - implications for breast cancer invasion. J Cell Sci. 2024;137:jcs261145.
[Google Scholar]
[CrossRef]
|
| 160. |
Xue C, Gao Y, Li X, Zhang M, Yang Y, Han Q, et al. Mesenchymal stem cells derived from adipose accelerate the progression of colon cancer by inducing a MT-CAFs phenotype via TRPC3/NF-KB axis. Stem Cell Res Ther. 2022;13:335.
[Google Scholar]
[CrossRef]
|
| 161. |
Dornen J, Myklebost O, Dittmar T. Cell Fusion of Mesenchymal Stem/Stromal Cells and Breast Cancer Cells Leads to the Formation of Hybrid Cells Exhibiting Diverse and Individual (Stem Cell) Characteristics. Int J Mol Sci. 2020;21:9636.
[Google Scholar]
[CrossRef]
|
| 162. |
Zhang LN, Kong CF, Zhao D, Cong XL, Wang SS, Ma L, et al. Fusion with mesenchymal stem cells differentially affects tumorigenic and metastatic abilities of lung cancer cells. J Cell Physiol. 2019;234:3570-3582.
[Google Scholar]
[CrossRef]
|
| 163. |
He X, Li B, Shao Y, Zhao N, Hsu Y, Zhang Z, et al. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer. 2015;15:24.
[Google Scholar]
[CrossRef]
|
| 164. |
Sun C, Dai X, Zhao D, Wang H, Rong X, Huang Q, et al. Mesenchymal stem cells promote glioma neovascularization in vivo by fusing with cancer stem cells. BMC Cancer. 2019;19:1240.
[Google Scholar]
[CrossRef]
|
| 165. |
Samanta S, Rajasingh S, Drosos N, Zhou Z, Dawn B, Rajasingh J. Exosomes: new molecular targets of diseases. Acta Pharmacol Sin. 2018;39:501-513.
[Google Scholar]
[CrossRef]
|
| 166. |
Lee YJ, Shin KJ, Chae YC. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp Mol Med. 2024;56:877-889.
[Google Scholar]
[CrossRef]
|
| 167. |
Han QF, Li WJ, Hu KS, Gao J, Zhai WL, Yang JH, et al. Exosome biogenesis: machinery, regulation, and therapeutic implications in cancer. Mol Cancer. 2022;21:207.
[Google Scholar]
[CrossRef]
|
| 168. |
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367.
[Google Scholar]
[CrossRef]
|
| 169. |
Chen YF, Luh F, Ho YS, Yen Y. Exosomes: a review of biologic function, diagnostic and targeted therapy applications, and clinical trials. J Biomed Sci. 2024;31:67.
[Google Scholar]
[CrossRef]
|
| 170. |
Yang X, Liu R, Jin J, Xv J, Wu J, Jin Y, et al. Cancer stem cells-derived exosomal TSPAN8 enhances non-stem cancer cells stemness and promotes malignant progression in PDAC. Oncogene. 2025;44:2328-2341.
[Google Scholar]
[CrossRef]
|
| 171. |
Li L, Zhao J, Zhang Q, Tao Y, Shen C, Li R, et al. Cancer Cell-Derived Exosomes Promote HCC Tumorigenesis Through Hedgehog Pathway. Front Oncol. 2021;11:756205.
[Google Scholar]
[CrossRef]
|
| 172. |
Li W, Zhang L, Guo B, Deng J, Wu S, Li F, et al. Exosomal FMR1-AS1 facilitates maintaining cancer stem-like cell dynamic equilibrium via TLR7/NFkappaB/c-Myc signaling in female esophageal carcinoma. Mol Cancer. 2019;18:22.
[Google Scholar]
[CrossRef]
|
| 173. |
Sun Z, Wang L, Zhou Y, Dong L, Ma W, Lv L, et al. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell Mol Neurobiol. 2020;40:767-784.
[Google Scholar]
[CrossRef]
|
| 174. |
Pan G, Jiang B, Yi Z, Yin J, Liu Y. Exosomal miR-105-5p derived from bladder cancer stem cells targets for GPR12 to promote the malignancy of bladder cancer. BMC Urol. 2023;23:155.
[Google Scholar]
[CrossRef]
|
| 175. |
Wang L, He M, Fu L, Jin Y. Exosomal release of microRNA-454 by breast cancer cells sustains biological properties of cancer stem cells via the PRRT2/Wnt axis in ovarian cancer. Life Sci. 2020;257:118024.
[Google Scholar]
[CrossRef]
|
| 176. |
Li X, Liu D, Chen H, Zeng B, Zhao Q, Zhang Y, et al. Melanoma stem cells promote metastasis via exosomal miR-1268a inactivation of autophagy. Biol Res. 2022;55:29.
[Google Scholar]
[CrossRef]
|
| 177. |
Liu D, Li X, Zeng B, Zhao Q, Chen H, Zhang Y, et al. Exosomal microRNA-4535 of Melanoma Stem Cells Promotes Metastasis by Inhibiting Autophagy Pathway. Stem Cell Rev Rep. 2023;19:155-169.
[Google Scholar]
[CrossRef]
|
| 178. |
Wang L, He J, Hu H, Tu L, Sun Z, Liu Y, et al. Lung CSC-derived exosomal miR-210-3p contributes to a pro-metastatic phenotype in lung cancer by targeting FGFRL1. J Cell Mol Med. 2020;24:6324-6339.
[Google Scholar]
[CrossRef]
|
| 179. |
Wang L, Yang G, Zhao D, Wang J, Bai Y, Peng Q, et al. CD103-positive CSC exosome promotes EMT of clear cell renal cell carcinoma: role of remote MiR-19b-3p. Mol Cancer. 2019;18:86.
[Google Scholar]
[CrossRef]
|
| 180. |
Hou ZH, Xu XW, Fu XY, Zhou LD, Liu SP, Tan DM. Long non-coding RNA MALAT1 promotes angiogenesis and immunosuppressive properties of HCC cells by sponging miR-140. Am J Physiol Cell Physiol. 2020;318:C649-C663.
[Google Scholar]
[CrossRef]
|
| 181. |
Shi L, Li B, Tan J, Zhu L, Zhang S, Zhang Y, et al. Exosomal lncRNA Mir100hg from lung cancer stem cells activates H3K14 lactylation to enhance metastatic activity in non-stem lung cancer cells. J Nanobiotechnology. 2025;23:156.
[Google Scholar]
[CrossRef]
|
| 182. |
Guo Y, Cui J, Liang X, Chen T, Lu C, Peng T. Pancreatic cancer stem cell-derived exosomal miR-210 mediates macrophage M2 polarization and promotes gemcitabine resistance by targeting FGFRL1. Int Immunopharmacol. 2024;127:111407.
[Google Scholar]
[CrossRef]
|
| 183. |
Wu L, Ye S, Yao Y, Zhang C, Liu W. Oral Cancer Stem Cell-Derived Small Extracellular Vesicles Promote M2 Macrophage Polarization and Suppress CD4(+) T-Cell Activity by Transferring UCA1 and Targeting LAMC2. Stem Cells Int. 2022;2022:5817684.
[Google Scholar]
[CrossRef]
|
| 184. |
Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, et al. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology. 2018;7:e1478647.
[Google Scholar]
[CrossRef]
|
| 185. |
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7:e1412909.
[Google Scholar]
[CrossRef]
|
| 186. |
Hwang WL, Lan HY, Cheng WC, Huang SC, Yang MH. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J Hematol Oncol. 2019;12:10.
[Google Scholar]
[CrossRef]
|
| 187. |
Huang YJ, Huang TH, Yadav VK, Sumitra MR, Tzeng DT, Wei PL, et al. Preclinical investigation of ovatodiolide as a potential inhibitor of colon cancer stem cells via downregulating sphere-derived exosomal beta-catenin/STAT3/miR-1246 cargoes. Am J Cancer Res. 2020;10:2337-2354.
[Google Scholar]
|
| 188. |
Grange C, Tapparo M, Tritta S, Deregibus MC, Battaglia A, Gontero P, et al. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer. 2015;15:1009.
[Google Scholar]
[CrossRef]
|
| 189. |
Chen JH, Wu ATH, Bamodu OA, Yadav VK, Chao TY, Tzeng YM, et al. Ovatodiolide Suppresses Oral Cancer Malignancy by Down-Regulating Exosomal Mir-21/STAT3/beta-Catenin Cargo and Preventing Oncogenic Transformation of Normal Gingival Fibroblasts. Cancers (Basel). 2019;12:56.
[Google Scholar]
[CrossRef]
|
| 190. |
Yang Z, Zhao N, Cui J, Wu H, Xiong J, Peng T. Exosomes derived from cancer stem cells of gemcitabine-resistant pancreatic cancer cells enhance drug resistance by delivering miR-210. Cell Oncol (Dordr). 2020;43:123-136.
[Google Scholar]
[CrossRef]
|
| 191. |
Huang H, Hou J, Liu K, Liu Q, Shen L, Liu B, et al. RAB27A-dependent release of exosomes by liver cancer stem cells induces Nanog expression in their differentiated progenies and confers regorafenib resistance. J Gastroen Hepatol. 2021;36:3429-3437.
[Google Scholar]
[CrossRef]
|
| 192. |
Koh MZ, Ho WY, Yeap SK, Ali NM, Yong CY, Boo L, et al. Exosomal-microRNA transcriptome profiling of Parental and CSC-like MDA-MB-231 cells in response to cisplatin treatment. Pathol Res Pract. 2022;233:153854.
[Google Scholar]
[CrossRef]
|
| 193. |
Shen M, Dong C, Ruan X, Yan W, Cao M, Pizzo D, et al. Chemotherapy-Induced Extracellular Vesicle miRNAs Promote Breast Cancer Stemness by Targeting ONECUT2. Cancer Res. 2019;79:3608-3621.
[Google Scholar]
[CrossRef]
|
| 194. |
Zhang H, Wang M, He Y, Deng T, Liu R, Wang W, et al. Chemotoxicity-induced exosomal lncFERO regulates ferroptosis and stemness in gastric cancer stem cells. Cell Death Dis. 2021;12:1116.
[Google Scholar]
[CrossRef]
|
| 195. |
Chung WM, Molony RD, Lee YF. Non-stem bladder cancer cell-derived extracellular vesicles promote cancer stem cell survival in response to chemotherapy. Stem Cell Res Ther. 2021;12:533.
[Google Scholar]
[CrossRef]
|
| 196. |
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932-3948.
[Google Scholar]
[CrossRef]
|
| 197. |
Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene. 2019;38:1951-1965.
[Google Scholar]
[CrossRef]
|
| 198. |
Wang Y, Yin K, Tian J, Xia X, Ma J, Tang X, et al. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv Sci (Weinh). 2019;6:1901278.
[Google Scholar]
[CrossRef]
|
| 199. |
Wang Y, Liu H, Zhang Z, Bian D, Shao K, Wang S, et al. G-MDSC-derived exosomes mediate the differentiation of M-MDSC into M2 macrophages promoting colitis-to-cancer transition. J Immunother Cancer. 2023;11.
[Google Scholar]
[CrossRef]
|
| 200. |
Li H, Li F. Exosomes from BM-MSCs increase the population of CSCs via transfer of miR-142-3p. Br J Cancer. 2018;119:744-755.
[Google Scholar]
[CrossRef]
|
| 201. |
Lyu T, Wang Y, Li D, Yang H, Qin B, Zhang W, et al. Exosomes from BM-MSCs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance via upregulation of S100A4. Exp Hematol Oncol. 2021;10:24.
[Google Scholar]
[CrossRef]
|
| 202. |
Yao X, Mao Y, Wu D, Zhu Y, Lu J, Huang Y, et al. Exosomal circ_0030167 derived from BM-MSCs inhibits the invasion, migration, proliferation and stemness of pancreatic cancer cells by sponging miR-338-5p and targeting the Wif1/Wnt8/beta-catenin axis. Cancer Lett. 2021;512:38-50.
[Google Scholar]
[CrossRef]
|
| 203. |
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69-82.
[Google Scholar]
[CrossRef]
|
| 204. |
Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23:171-185.
[Google Scholar]
[CrossRef]
|
| 205. |
McCoy MG, Nyanyo D, Hung CK, Goerger JP, W RZ, Williams RM, et al. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci Rep. 2019;9:9069.
[Google Scholar]
[CrossRef]
|
| 206. |
Krishnamurthy S, Warner KA, Dong Z, Imai A, Nor C, Ward BB, et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells. 2014;32:2845-2857.
[Google Scholar]
[CrossRef]
|
| 207. |
Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F, et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer. 2015;14:155.
[Google Scholar]
[CrossRef]
|
| 208. |
McGettrick AF, O’Neill LAJ. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020;32:524-536.
[Google Scholar]
[CrossRef]
|
| 209. |
Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014;49:1-15.
[Google Scholar]
[CrossRef]
|
| 210. |
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343-354.
[Google Scholar]
[CrossRef]
|
| 211. |
Zimna A, Kurpisz M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res Int. 2015;2015:549412.
[Google Scholar]
[CrossRef]
|
| 212. |
Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8:70.
[Google Scholar]
[CrossRef]
|
| 213. |
Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102:789-795.
[Google Scholar]
[CrossRef]
|
| 214. |
Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int. 2021;21:62.
[Google Scholar]
[CrossRef]
|
| 215. |
Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617-628.
[Google Scholar]
[CrossRef]
|
| 216. |
Wang P, Wan WW, Xiong SL, Feng H, Wu N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017;3:16105.
[Google Scholar]
[CrossRef]
|
| 217. |
Seo EJ, Kim DK, Jang IH, Choi EJ, Shin SH, Lee SI, et al. Hypoxia-NOTCH1-SOX2 signaling is important for maintaining cancer stem cells in ovarian cancer. Oncotarget. 2016;7:55624-55638.
[Google Scholar]
[CrossRef]
|
| 218. |
Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012;19:284-294.
[Google Scholar]
[CrossRef]
|
| 219. |
Dong HJ, Jang GB, Lee HY, Park SR, Kim JY, Nam JS, et al. The Wnt/beta-catenin signaling/Id2 cascade mediates the effects of hypoxia on the hierarchy of colorectal-cancer stem cells. Sci Rep. 2016;6:22966.
[Google Scholar]
[CrossRef]
|
| 220. |
Boso D, Rampazzo E, Zanon C, Bresolin S, Maule F, Porcu E, et al. HIF-1alpha/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics. 2019;9:4860-4877.
[Google Scholar]
[CrossRef]
|
| 221. |
Bijlsma MF, Groot AP, Oduro JP, Franken RJ, Schoenmakers S, Peppelenbosch MP, et al. Hypoxia induces a hedgehog response mediated by HIF-1alpha. J Cell Mol Med. 2009;13:2053-2060.
[Google Scholar]
[CrossRef]
|
| 222. |
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776-779.
[Google Scholar]
[CrossRef]
|
| 223. |
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165-172.
[Google Scholar]
[CrossRef]
|
| 224. |
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer. 2013;12:66.
[Google Scholar]
[CrossRef]
|
| 225. |
Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ, et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013;73:3235-3247.
[Google Scholar]
[CrossRef]
|
| 226. |
Hallis SP, Kim SK, Lee JH, Kwak MK. Association of NRF2 with HIF-2alpha-induced cancer stem cell phenotypes in chronic hypoxic condition. Redox Biol. 2023;60:102632.
[Google Scholar]
[CrossRef]
|
| 227. |
Wang P, Gong S, Liao B, Pan J, Wang J, Zou D, et al. HIF1alpha/HIF2alpha induces glioma cell dedifferentiation into cancer stem cells through Sox2 under hypoxic conditions. J Cancer. 2022;13:1-14.
[Google Scholar]
[CrossRef]
|
| 228. |
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2alpha targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 2022;29:1769-1789.
[Google Scholar]
[CrossRef]
|
| 229. |
Ramakrishnan S, Anand V, Roy S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol. 2014;9:142-160.
[Google Scholar]
[CrossRef]
|
| 230. |
Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34:3107-3119.
[Google Scholar]
[CrossRef]
|
| 231. |
Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. GLI1 regulates a novel neuropilin-2/alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med. 2013;5:488-508.
[Google Scholar]
[CrossRef]
|
| 232. |
Xu Z, Goel HL, Burkart C, Burman L, Chong YE, Barber AG, et al. Inhibition of VEGF binding to neuropilin-2 enhances chemosensitivity and inhibits metastasis in triple-negative breast cancer. Sci Transl Med. 2023;15:eadf1128.
[Google Scholar]
[CrossRef]
|
| 233. |
Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauss A, et al. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol. 2020;11:1280.
[Google Scholar]
[CrossRef]
|
| 234. |
Spit M, Koo BK, Maurice MM. Tales from the crypt: intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol. 2018;8.
[Google Scholar]
[CrossRef]
|
| 235. |
Zhang B, Chen T. Local and systemic mechanisms that control the hair follicle stem cell niche. Nat Rev Mol Cell Biol. 2024;25:87-100.
[Google Scholar]
[CrossRef]
|
| 236. |
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327-334.
[Google Scholar]
[CrossRef]
|
| 237. |
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
[Google Scholar]
[CrossRef]
|
| 238. |
Izadpanah A, Mohammadkhani N, Masoudnia M, Ghasemzad M, Saeedian A, Mehdizadeh H, et al. Update on immune-based therapy strategies targeting cancer stem cells. Cancer Med. 2023;12:18960-18980.
[Google Scholar]
[CrossRef]
|

 ,
Na Li
1
,
Na Li
1